
石墨炉原子吸收法测定水产品铅检出限和不确定度及其应用探讨
2020-10-28 09:20李兆千
中国渔业质量与标准 2020年5期
李兆千
(天津市农业生态环境监测与农产品质量检测中心,天津 300221)
不确定度是指由于测量误差的存在,对被测量值的不能肯定的程度。目前不确定度评定已广泛应用于检测方法的选择、检测水平的评估以及不确定度降低等方面。如用于指导医学工程部门进行除颤仪质量控制[1]、解决测绘技术中平差算法中不确定度的选择问题[2]和分析降低锅炉热效率测试不确定度的方法等[3]。
目前,中国水产品中铅检出存在超标率现象。2017年在南通市区范围内4类440份水产品检测中,梭子蟹(Portunustrituberculatus)超标率最高,为9.1%[4];2010—2014年广东省共采集市售水产品样品1 326份,检测出铅含量均值为0.040 mg/kg,超标率为0.15%[5]。
《食品安全国家标准 食品中铅的测定》(GB 5009.12—2017)[6]、《食品安全国家标准 食品中多元素的测定》(GB 5009.268—2016)[7]和《食品安全国家标准 食品中镉的测定》(GB 5009.15—2014)[8]的检出限以“mg/kg”为单位,但当测定一个批次样品时,由于质量、稀释体积不一样,以“mg/kg”为单位的检出限并不能统一表示。因此,改变检出限的表示形式,对统一检出限有重要的意义。《食品安全国家标准 食品中铅的测定》(GB 5009.12—2017)规定精密度:在重复性条件下,获得的两次独立测定结果的绝对差值不得超过算术平均值的20%[5],按精密度的计算方法,将不确定度的两极值带入公式,发现部分研究结果超过了规定的精密度。如测定猫须草(Clerodendranthusspicatus)[9]、水产品[10]、乳[11]、阿胶[12]、烘焙食品[13]和条斑紫菜(Porphyrayezoensis)[14]中铅的不确定度分别为:(2.28±0.34)、(0.33±0.16)、(0.040±0.007 6)、(0.466 0±0.056 9)、(0.320±0.168)和(0.440±0.054) mg/kg,两极值的精密度分别为:29.8%、97.0%、38.0%、24.4%、105.0%和24.5%。部分文献报道的在20%以内,如大米[15]、文蛤(Meretrixmeretrix)[16]、龟苓膏粉[17]和木薯[18]中铅的测量不确定度分别为:(0.100±0.009)、(0.780±0.032 3)、(0.902±0.064)和(0.058 00±0.000 45) mg/kg,两极值的精密度分别为:18.0%、14.2%、8.3%和1.6%。因此,改变标准的精密度要求对规范检测能力有重要意义。
本研究参考《化学分析测量不确定度评定》[19]、《测量不确定度评定与表示》[20]以及检出限的测量相关文献,进行水产品中铅含量的不确定度评定, 为评价实验操作、提高检测结果准确度提供参考依据。
1 材料与方法
1.1 材料与试剂
实验用100 μg/mL铅标准溶液GBW(E)080129(中国计量科学研究院批号14061),相对扩展不确定度为(100.0±0.8) μg/mL (k=2),使用前应恒温至(20±5) ℃;1 000 mg/L硝酸钯(PerkinElmer公司);硝酸(高恒批号161206 BⅤ-Ⅲ,北京化学试剂研究所);磷酸二氢铵(优级纯,天津市科密欧化学试剂有限公司);实验用水符合二级水规格[21]。
1.2 仪器与设备
ZA3000原子吸收光谱仪购置于天美仪器有限公司。PRACTUM213-1CN电子天平购置于赛多利斯(最大允许误差为5 mg)。移液枪购置于Eppendorf公司。
1.3 方法
铅标准使用液的配制:将铅标准品溶液在(20±5) ℃放置10 min。摇匀后用1 mL移液枪准确吸取1.00 mL铅标准贮备溶液至10 mL容量瓶中,用1%硝酸定容,得到质量浓度为10 mg/L铅标准中间液。后用100 μL移液器分别准确吸取50 μL至10 mL容量瓶中,用1%硝酸定容,混匀后为50 μg/L标准使用液。
样品前处理:按照GB 5009.12—2017第一法中“微波消解”法进行前处理。如超过标准溶液线性范围,对样品稀释处理。
1.4 建立不确定度数学模型
试样中铅的含量按式(1)计算。
式(1)
式(1)中:X代表试样中铅的含量,单位为mg/kg或mg/L;C代表试样溶液中铅的质量浓度,单位为μg/L;V代表试样消化液的定容体积,单位为mL;m代表试样称样量或移取体积,单位为g或mL;Ck代表样品空白浓度,单位为μg/L;换算系数为1 000。
1.5 不确定度来源分析
据方法数学模型分析,不确定度来源主要有:标准曲线最高测量点相对不确定度Urel(B)、标准曲线拟合相对不确定度Urel(Q)、平行样测量及重复测量相对不确定度Urel(C)、空白测量相对不确定度Urel(Ck)、试样称量相对不确定度Urel(m)和回收率相对不确定度Urel(R)。
仪器条件参数包括:峰高峰面积的选择、灰化温度、原子化温度、灯电流、样品加入体积及基体改进剂加入体积等。经调查发现,以仪器参数作为不确定度的论文文献甚少,一般通过将仪器调节到最佳条件,分析上述数学模型的不确定度。本研究中,同样不对上述仪器条件参数做评定。
2 各分量相对不确定度的计算
2.1 由标准曲线最高点产生的相对不确定度
2.1.1 标准储备液定值引入的相对不确定度
依据标准物质证书,100 μg/mL铅标准溶液GBW(E)080129(批号14061)的相对扩展不确定度为0.8% (k=2),标准储备液定值引入的相对不确定度见式(2)。
式(2)
式(2)中:u(c标储)代表相对扩展不确定度;C标储代表标准溶液浓度;k代表包含因子。
2.1.2 配置标准曲线最高点引入的相对不确定度
1)容量瓶引起的不确定度
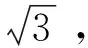
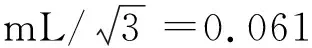
③第一次稀释至10 mL容量瓶产生的相对不确定度见式(3)。
式(3)
式(3)中:u(V10)代表不确定度;V10代表溶液体积。
④第二次稀释至10 mL容量瓶产生的相对不确定度见式(4)。
式(4)
2)移液枪引起的相对不确定度
移液器已经过JJG 646—2006《移液器检定规程》[23]检定。
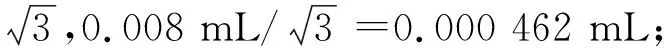
③1 mL移液枪产生的体积相对不确定度见式(5)。
式(5)
式(5)中:u(V枪1)代表不确定度;V(枪1)代表溶液体积。
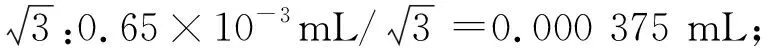
式(6)
式(6)中:u(V枪2)代表不确定度;V枪2代表溶液体积。
综上,标准溶液产生的相对不确定度见式(7)。

式(7)
2.2 天平称量误差引入的相对不确定度

式(8)
式(8)中:Urel(m)代表相对不确定度,u(m)代表不确定度;m代表质量。
2.3 标准曲线拟合引入的相对不确定度
实验配置40 μg/mL的标准溶液,仪器将标准溶液分别配制0、8、16、24、32及40 μg/mL的注射液,用石墨炉原子吸收光谱仪测定5次,得到相应的吸光度,用最小二乘法进行拟合,得到线性回归方程y=a+bc及相关系数(r)。表1为铅标准曲线测定结果。
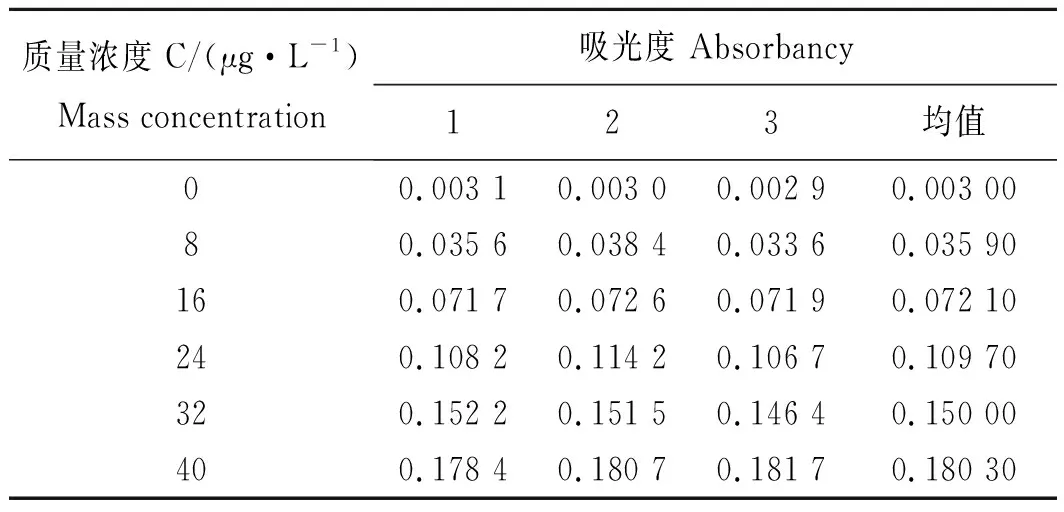
表1 铅标准曲线测定结果Tab.1 Determination result of lead standard curve
标准曲线回归方程:y=0.004 58x-0.000 21,r=0.999 53。
根据贝塞尔公式,试验标准差见式(9)。
式(9)
式(9)中:S代表试验标准偏差;yi代表对应质量浓度的单次吸光度;a代表截距;b代表斜率;ci代表质量浓度;n代表总测量次数,即18次。
标准曲线拟合引入的相对不确定度见式(10)。
式(10)
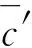
2.4 样品重复测定引入的相对不确定度
以拟合的标准曲线为定量标准,对样品进行了4次测量,检测浓度分别为4.683 1、5.607 6、5.525 7和6.624 3μg/L。4次重复测定的平均值为5.610 2 μg/L,则样品重复测定引入的相对不确定度见式(11)
式(11)
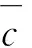
2.5 平行样测定引入的相对不确定度
以拟合的标准曲线为定量标准,对5个平行样品进行了测量,得到样品中铅元素检测浓度分别为5.610 2、6.721 4、6.065 4、8.057 1和7.426 9μg/L,定容体积为10 mL,平行样质量分别为0.298、0.332、0.307、0.363和0.323 g,经式(1)计算,质量浓度分别为0.188 3、0.202 5、0.197 6、0.222 0和0.229 9 mg/kg。
5个平行样的平均值为0.208 1 mg/kg,则样品平行样测量相对标准不确定度见式(12)。
0.037 31
式(12)
2.6 试剂空白的相对不确定度
检测的未扣除空白值的试剂空白值为:1.318 2、1.011 7、1.423 4、1.181 0、1.456 8、0.971 9、1.165 3、0.962 5、1.164 1和0.872 7 μg/L。
10个试剂空白的平均值为1.153 μg/L,则试剂空白平行样的测量相对标准不确定度见式(13)
式 (13)

2.7 加标回收引入的相对不确定度[24]
样品加标回收法可以度量方法偏差及其不确定度。方法偏差SF的不确定度U(SF)评估模型见式(14)
式(14)
2.7.1 加标物质与实际物质行为差异f行为引入的不确定度U(f行为)
式(15)

式(16)

式(17)
式(17)中:n加标代表加标样品测定个数;n样代表测定样品的个数。
式(18)

差异的显著性检验:
式(19)
式(20)
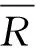
所用的标准样品标准值为0.20 mg/kg,加标质量1×10-4mg,溶液体积10.00 mL,试剂空白浓度1.043 9 μg/L。样品及加标信息、方法偏差试验结果见表2、表3。

表2 标准样品信息Tab.2 Standard sample information

表3 加标回收样品信息Tab.3 Sample information on standard addition recovery test
3 相对不确定度组成图及控制标准曲线相对不确定度分量的方法
总结出定容不确定度,标液不确定度、曲线拟合不确定度、空白测定不确定度及样品重复测定相对不确定度见表4。
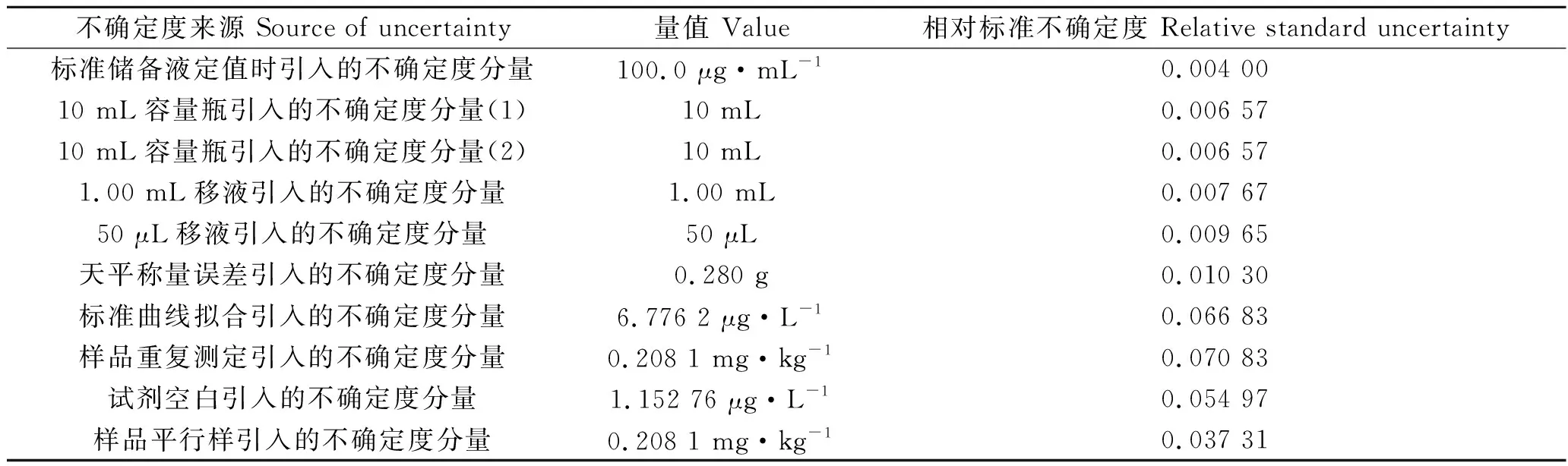
表4 相对不确定度分量一览表Tab.4 Uncertainty components list
3.1 相对不确定度组成占比图
将容量瓶、移液器引起的不确定度合并为配线过程不确定度,将样品重复测定和平行样引起的不确定度合并为重复测定不确定度。不确定度分量占不确定度总量的比例见图1。
从图1可知,对检测影响最大的因素是重复性测定不确定度分量,占比为39.37%;其次是曲线拟合不确定度分量,占比24.33%;试剂空白不确定度分量,占比总计20.01%;配置标准曲线和质量部分不确定度分量总计占比11.09%。
3.2 减小相对不确定度分量的方法
曲线拟合、重复测定和试剂空白不确定度是不确定度的主要因素。
可以通过以下两种方法降低不确定度。一是设定曲线量程法,根据预测的样品值设定曲线,使样品测定值处在标准曲线正中间。二是增测次数法,增加样品和标准曲线的测量次数,减小不确定度分量。
式(21)
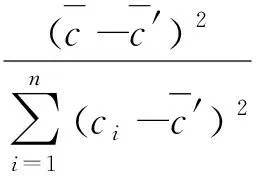
4 检出限
12个样品空白平行样的吸光度分别为:0.005 8、0.004 4、0.006 3、0.005 2、0.006 5、0.004 2、0.005 1、0.004 2、0.005 1和0.003 8。
式(22)
式(23)
DEL代表检出限,k代表曲线斜率,经计算检出限为1.196 μg/L,当称样量为0.5 g,定容体积为10 mL时,质量浓度的检出限为0.023 92 mg/kg。
5 扩展不确定度
综上,微波消解-石墨炉原子吸收光谱法测定水产品中铅含量的相对不确定度Urel(w)见式(22)。
Urel(w)=
0.148 5
式(22)
计算其合成不确定度为:
u(w)=0.208 1mg×0.148 5=0.030 9 mg/kg
式(23)
取包含因子k=2(95%置信概率),则铅的扩展不确定度为:
U=k×u(w)=2×0.030 9=0.061 mg/kg
式(24)
经计算,该样品不确定度表示为:(0.208±0.061) mg/kg。
6 结论
6.1 检出限
得到检测溶液的检出限为1.196 μg/L,当称样量为0.5 g,定容体积为10 mL时,样品的检出限为0.023 92 mg/kg。与溶液检出限相关的参数包括2个变量,空白相对标准偏差和曲线斜率,是测定一批次样品时不变的参数。而以样品检出限相关的参数增加了2个变量,称样量和体积,在一批次测定中参数不一致。所以,以“μg/L”为单位可以统一一批次测定量的检出限。
6.2 精密度要求
标准中规定的精密度要求较小。标准中规定:在重复性条件下获得的两次独立测定结果的绝对差值不得超过算术平均值的20%。而在许多报道的文献中,不确定度两极值的精密度超过了标准规定的20%。主要有以下原因:1)未对数量级分级进行精密度要求;2)有的种类的样品测定的干扰度大,相对标准偏差大。
6.3 降低相对标准偏差的方法
由于调整曲线量程法不能满足实际检测过程要求,所以降低测量不确定度的方法是在允许的情况下对曲线和平行样品进行多次测量。
猜你喜欢
——以离子色谱法测定冬瓜中亚硝酸盐含量为例
现代食品(2022年15期)2022-09-08
波谱学杂志(2022年2期)2022-06-14
系统医学(2022年6期)2022-06-13
数学大王·低年级(2022年5期)2022-05-21
四川农业科技(2021年9期)2021-11-19
高中生学习·高二版(2017年9期)2017-10-25
科学家(2016年17期)2017-10-17
幼儿智力世界(2016年11期)2017-02-21
科技视界(2016年22期)2016-10-18
科技与创新(2015年24期)2015-12-21
