
仔猪腹泻病原菌多重PCR检测方法的建立
2020-10-15 14:39申湖黄利
兽医导刊 2020年16期
申 湖 黄 利
(大方县农业农村局,贵州大方 551600)
1 材料
1.1 菌株
大肠杆菌、沙门氏菌、志贺杆菌三种细菌均由贵州省动物疫病研究室提供。
1.2 试剂
普通营养琼脂培养基、鲜血营养琼脂培养基、麦康凯琼脂培养基、伊红美蓝培养基、胆硫乳培养基、三糖铁培养基、LB液体培养基,购自杭州微生物试剂有限公司。
2 方法
2.1 试剂配制
2.1.1 固体培养基
称取胰蛋白胨10g;酵母提取物5g;NaCl 10g;琼脂粉15g依次放入锥形瓶中加入蒸馏水至1000ml,加热溶液至溶液沸腾。将锥形瓶包装好后放入高压灭菌锅中进行121℃高压灭菌30min,灭菌后将溶液倾倒无菌平板即可,制作好的平板置于4℃冰箱保存待用。
2.1.2 液体培养基
称取胰蛋白胨10g,酵母提取物5g,NaCl 10g,依次放入锥形瓶中加入蒸馏水至1000 ml,用电热炉加热至沸腾待容易呈清亮透明时即可,最后将锥形瓶包装好后放入121℃高压灭菌30min。
2.1.3 琼脂糖凝胶
称取1.5g琼脂糖于100ml锥形瓶中,向瓶内加入TBE至100ml,在电热炉上将其加热煮沸待溶液清亮透明时即可。待溶液冷却至50~60℃时加入7μl核酸染料混匀,将配制好的电泳凝胶溶液倾倒胶槽中,待其彻底冷却方可使用。
2.2 引物设计与合成
针对大肠埃希菌23S rRNA基因、沙门氏菌invA基因和志贺杆菌的ipaH基因,用 Premier 5.0软件设计上下游引物,引物由英俊公司合成。
2.3 细菌DNA提取
将实验室保存的已分离鉴定的大肠杆菌、沙门氏菌、志贺杆菌用接种环接种到新的LB液体培养基中,于37℃恒温振荡培养箱中培养,用于DNA提取。
(1)从保存的离心管中取新的离心管并标上大肠杆菌、沙门氏菌、志贺杆菌字样然 后从纯化培养过的大肠杆菌、沙门氏菌、志贺杆菌试管中分别取2ml菌液加入到标记的离心管内,10000.rpm离心1min然后用移液枪吸掉或直接倒掉上清液,得到菌体沉淀。
(2)用移液枪分别向所获得的菌体沉淀离心管中加入200μl缓冲液GA,利用振荡仪充分震荡混匀至菌体沉淀彻底悬浮。
(3)向离心管中分别加入20 μlproteinaseK溶液然后上下颠倒使溶液充分混匀。
(4)往离心管中加入220 μl缓冲液GB振荡15s,70℃放置10 min这时溶液应变清亮,简短离心以去除离心管管盖内壁的水珠。
(5)向离心管内各加入无水乙醇220 μl,利用微量震荡器充分振荡溶液使其混匀,此可能会出现絮状沉淀,为了去除管盖内壁的水珠可以用微量离心机离心。
(6)将上一步所获得的絮状沉淀及其溶液转移到三个吸附柱CB1、CB2、CB3中,并将吸附柱放入到收集管内,12000 rpm离心30s,倾倒掉废液并把吸附柱放回到收集管内
(7)向三支吸附柱中分别加入500 μl缓冲液GD,12000 rpm离心30s然后倒掉废液并把吸附柱重新放回收集管内
(8)往三支吸附柱中分别加入600 μl漂洗液PWC,12000 rpm离心30s,倾倒掉上层废液并把吸附柱放回收集管内
(9)继续步骤8
(10)将三支吸附柱放回收集管内,12000 rpm空柱离心2 min并倾倒掉废液,将三支吸附柱室温静置5 min。
(11)把三支吸附柱分别放入三支新的收集管中,向吸附柱中间部位悬空滴加50~200μl的洗脱缓冲液TE,室温静置5 min,然后12000 rpm离心2 min并将所得溶液收集到离心管内。

表2 引物序列表
2.4 多重PCR检测方法的建立
2.4.1 单重PCR检测
分别以大肠埃希菌、志贺杆菌和沙门氏菌菌株DNA为模板进行PCR扩增,总体积25μlPCR反应体系,结束后用1.2%的琼脂糖凝胶电泳。反应条件和体系见表3

表3 单重PCR反应体系
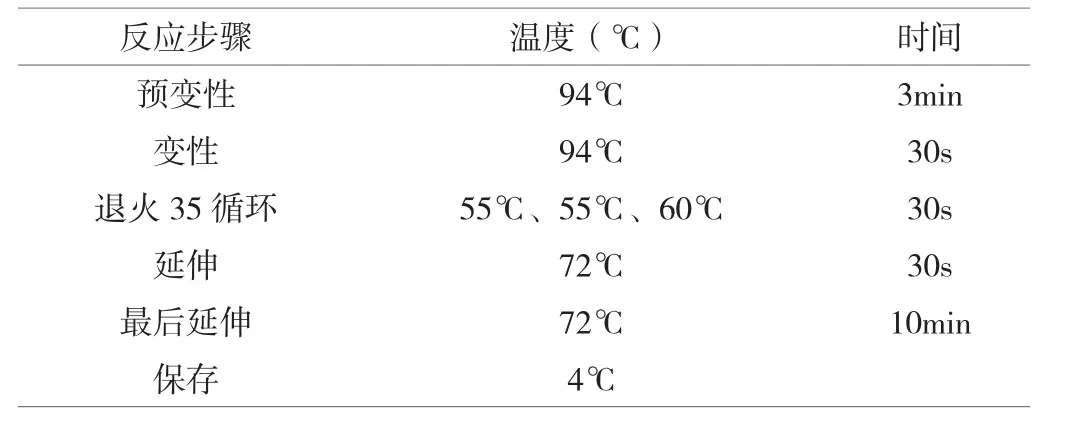
表4 单重PCR反应条件
2.4.2 多重PCR扩增
按照实验步骤2.4的方法提取大肠杆菌、沙门氏菌和志贺杆菌的DNA模版,将提取的三种细菌的DNA模版按大肠杆菌DNA:沙门氏菌DNA:志贺杆菌DNA=1:1:1等量混匀并以混匀的三种菌的DNA 作为模版进行目的基因的扩增反应体系见表5,反应条件见表6。

表5 单重PCR反应体系

表6 单重PCR反应条件
2.4.3 多重PCR反应引物浓度的优化(表7)
2.4.4 多重PCR反应退火温度的优化
应用上一步的最佳引物浓度和各退火温度组合对多重PCR退火温度进行优化,优化退火温度从51~60℃共10个梯度。从中优化出多重PCR反应体系的最佳退火温度。PCR扩增后经1.2%琼脂糖凝胶电泳。
2.4.5 多重PCR反应敏感性实验
利用步骤2.3提取的DNA模版经核酸蛋白测定仪对3种细菌基因组DNA样本进行浓度测定,并分别进行10倍梯度稀释,取各稀释度样本进行PCR扩增。检验多重PCR最佳灵敏度。
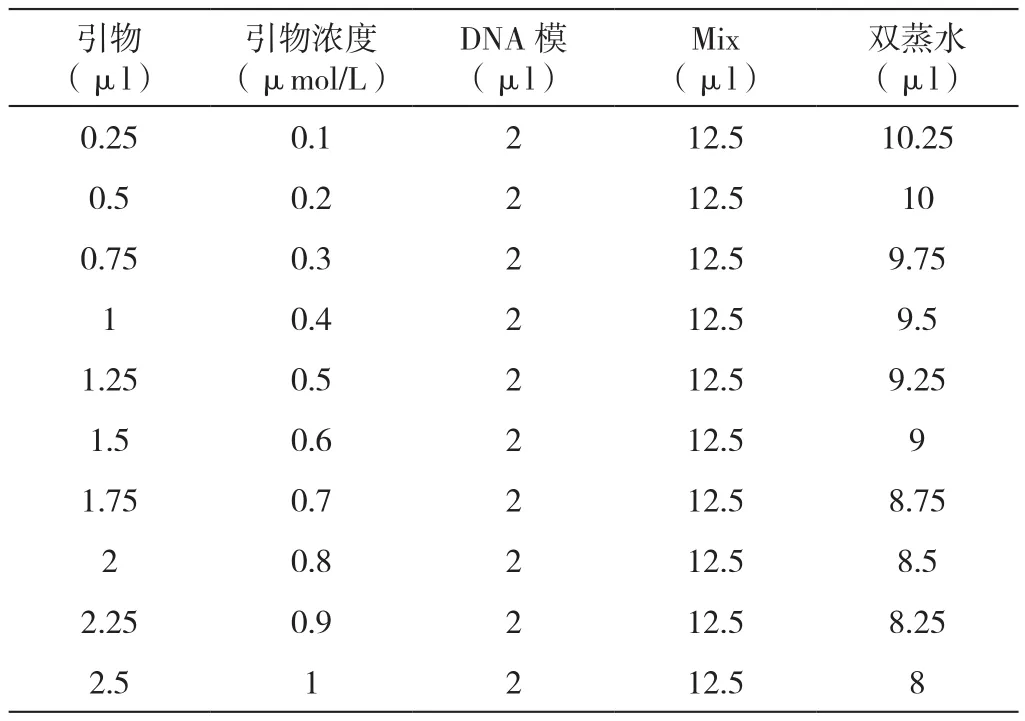
表7 引物浓度梯度表
2.4.6 多重PCR反应特异性实验
以大肠杆菌、沙门氏菌、志贺杆菌、葡萄球菌、链球菌、巴氏杆菌等细菌混合DNA为模板,同时加入3对引物进行扩增;同时加入3对引物进行PCR扩增。然后经1.2%的琼脂糖凝胶电泳观察
3 实验结果
3.1 单重PCR反应体系的建立
对大肠杆菌、沙门氏菌、志贺杆菌三种细菌单一模版进行单重PCR扩增。
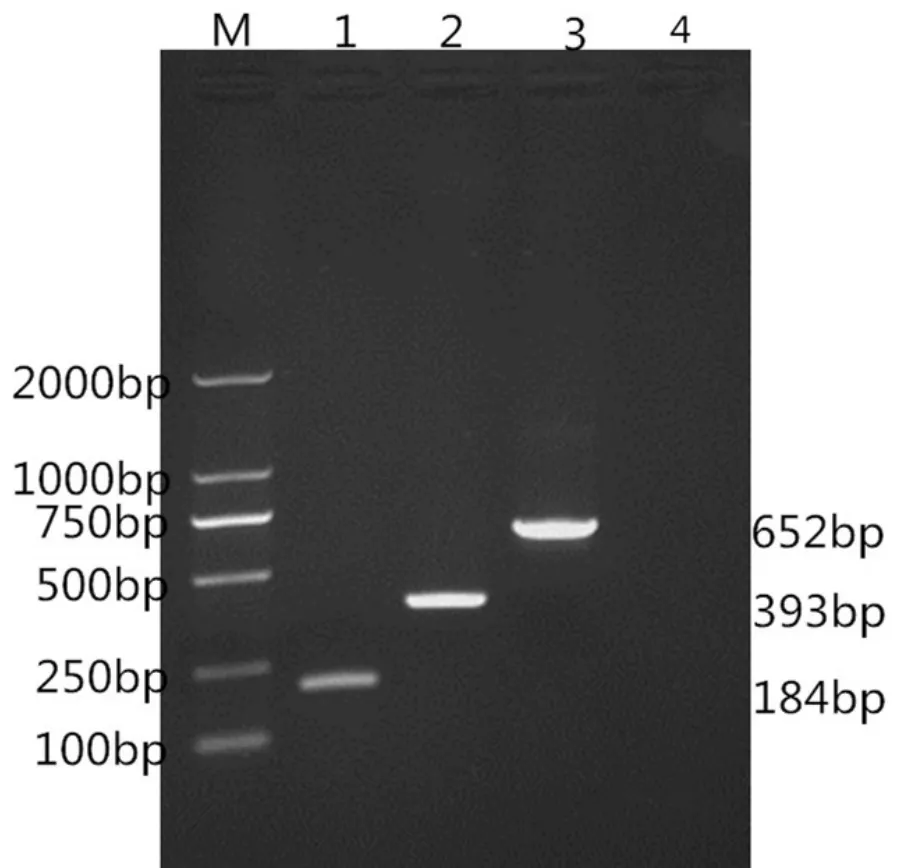
图1 单重PCR反应的建立
由图1可以看出大肠杆菌、沙门氏菌、志贺杆菌三种菌PCR扩增分别在652bp、184bp、393bp时得到清晰明亮的条带,说明,本次试验所研究设计的3对引物特异性较好,表明研究设计的三对引物能够应用到之后的多重PCR检测方法的建立。
3.2 多重PCR退火温度优化
选择51~60℃进行温度梯度PCR扩增,结果见图2
如图2所示,多重PCR均能得到清晰明亮目的条带,其中第5泳道的条带最明亮,因此选择55℃作为本研究试验的最佳退火温度。
3.3 多重PCR引物浓度优化
选择引物添加量分别为0.25μl、0.5μl、0.75μl、1μl、1.25μl、1.5μl、1.75μl、2μl、2.25μl、2.5μl,按所加引物浓度梯度进行PCR扩增,扩增结果见图3。
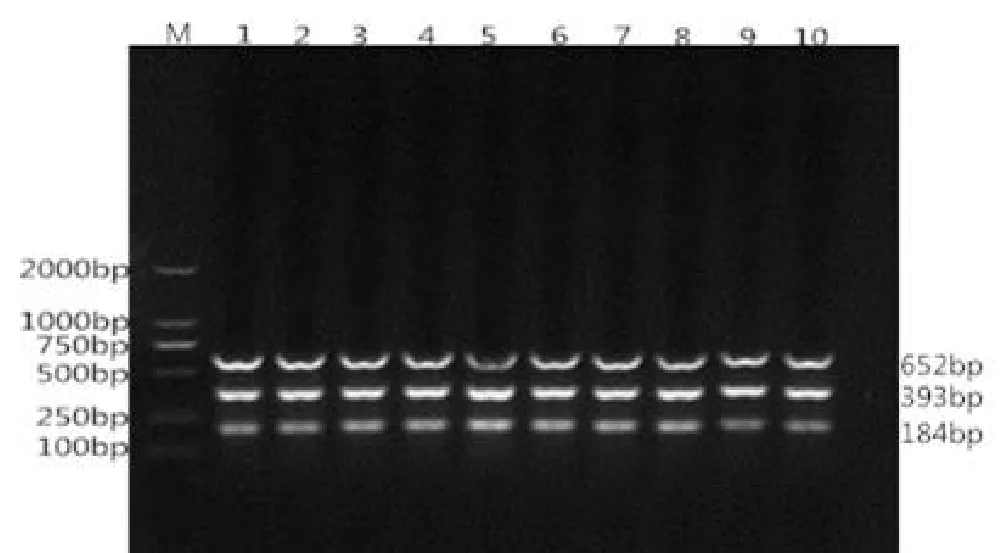
图2 多重PCR反应退火温度的优化
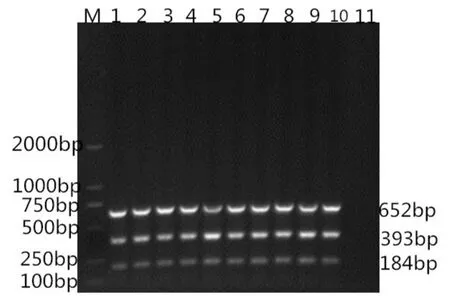
图3 多重PCR反应引物浓度的优化
如图3所示均得到目的条带但4泳道的条带最清晰因此选择引物添加量为1 μl即引物浓度为0.4 μmol/L作为最佳引物浓度
3.4 多重PCR敏感性试验
将初始三种DNA模板浓度分别稀释,然后跑三重PCR结果见图4。
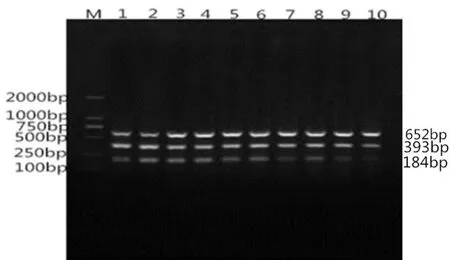
图4 多重PCR反应敏感性试验
由图4可见,设计模版稀释梯度为10-1、10-2、10-3、10-4、10-5、10-6、10-7、10-8、10-9。 图中第 8 泳道的条带最为暗淡,所以 多重PCR同时检测沙门氏菌、志贺氏菌和大肠杆菌灵敏度为10-8CFU/ml。
3.5 多重PCR特异性试验
分别对大肠杆菌、沙门氏菌、志贺杆菌3种细菌混合模版、单一模版及其他细菌的单一模版进行多重PCR扩增结果见图5。
特异性试验所建立多重PCR能够对3种细菌混合模版、单一模版扩增出清晰明亮的目的条带,未能扩增出其他细菌的任何目的条带,表明所建立的方法特异性良好。
4 讨论与分析
影响PCR反应的因素有很多,引物的特异性、稳定性、PCR反应引物的浓度、PCR体系的退火温度、DNA模版的含量和质量以及各个DNA模版之间相互影响等都会影响到检测的效果,因此可以从影响反应的因素出发,通过选择适当条件因素进行PCR反应来建立能够快速精确地检测出致病菌的检测方法。
而本次试验针对志贺杆菌ipaH的基因、大肠埃希菌23S rRNA基因和沙门氏菌invA基因分别设计合成了3对特异性引物。通过对大肠杆菌、沙门氏菌、志贺杆菌进行单重PCR扩增,在凝胶成像仪上可以看出设计的三对引物扩增的条带都比较清晰明亮,表明研究设计的三对引物具有较好的特异性和稳定性。通过不同的引物浓度梯度对模版DNA进行PCR扩增从中优化出最佳引物浓度,再用最佳引物浓度应用到多重PCR体系,选择不同的退火温度梯度对模版DNA进行PCR扩增,从而优化出最佳退火温度。通过对引物浓度及退火温度的优化极大的减少了PCR扩增误差,增加了实验结果的准确性。经过随机选取大肠杆菌、沙门氏菌、志贺杆菌等6种细菌菌株进行特异性实验,结果表明本研究试验设计的多重PCR检测方法具有良好的特异性和准确性。整个检测的过程所需时间极大缩短、准确度得到了很大的提高,这能够为疾病的控制和治疗节约大量的时间。

图5 多重PCR反应特异性试验
猜你喜欢
动物医学进展(2022年9期)2022-09-06
计算机应用与软件(2022年6期)2022-07-12
油气地质与采收率(2022年3期)2022-05-20
自然灾害学报(2022年2期)2022-05-10
中国饲料(2021年17期)2021-11-02
临床与实验病理学杂志(2021年7期)2021-09-06
食品安全导刊(2021年20期)2021-08-30
中国学校体育(2021年10期)2021-04-26
科技视界(2020年26期)2020-09-24
科技视界(2020年17期)2020-07-30
