
钯催化的7-氮杂吲哚的3位芳基化反应
2020-10-13 03:06:54刘珊珊周鲜颖马养民
陕西科技大学学报 2020年5期
刘珊珊,赵 晨,周鲜颖,马养民
(1.陕西科技大学 化学与化工学院,陕西 西安 710021;2.陕西科技大学 陕西省轻化工助剂重点实验室,陕西 西安 710021)
0 引言
7-氮杂吲哚骨架存在于天然产物Variolin衍生物的核心结构单元中,是很多药物分子的重要亚结构,具有抑制多种蛋白酶活性的功效,在抗组胺、抗多巴胺、抗菌、抗肿瘤等方面显示出潜在的药用价值[1,2].C3芳基化是合成Variolin B分子的重要步骤(如图1所示),以往的研究多是通过Suzuki或Stille偶联反应完成这一关键步骤的合成[3].而对底物的预先官能化成为实现这一反应必不可少的一步,不仅增加了合成成本,也会引入难以去除的杂质.因此,开发原子经济的合成路线实现这一反应成为化学家和药物学家追逐的目标.

图1 Variolin B的逆合成分析
近年来,7-氮杂吲哚选择性的官能化反应研究取得了一定的进展[4-9].早在2006年,Kerr等[10]在7-氮杂吲哚的C6位引入三氟甲磺酰基,通过Pd催化的偶联反应分别在目标位点实现了甲基化、芳基化、炔基化和酰基化转化.2016年,Das课题组研究了室温条件下7-氮杂吲哚在Pd催化下其C3位选择性的烯基化反应,在所有49个反应实例中,作者得到了高达85%的分离产率,值得注意的是,该反应全部发生在底物的C3位,表现出了极高的区域选择性[11].随后,该课题组以3-卤代-7-氮杂吲哚为底物,首次报道了以CHCl3为羰基源的7-氮杂吲哚酰胺化反应[12].最近,李帅帅等[13]系统地研究了Rh催化的7-氮杂吲哚串联环化反应,以30%~99%的收率合成了一系列7-氮杂吲哚衍生物,并说明该过程是由炔基化反应和随后的分子内C-H环化共同作用直接构建C-C键.
尽管目前关于7-氮杂吲哚骨架的修饰已经取得突破性进展,但引入芳基的方法依然较少.如图2所示,2009年,Fagnou等[14]报道了7-氮杂吲哚的选择性芳基化反应,作者以Pd(OAc)2为催化剂,首先通过N-甲基-7-氮杂吲哚与碘苯的反应,选择性地实现C2的直接芳基化反应;当作者引入N-O作为导向基团时,反应的区域选择性发生了转变,成功实现了C6选择性芳基化反应.2013年,Kannaboina等[15]以85%的产率实现了室温下Pd催化的N-甲基-7-氮杂吲哚与芳基硼酸的C2芳基化反应,继而以3-碘-5-溴-7-氮杂吲哚为起始物,通过与芳基硼酸的两次Suzuki偶联反应合成C2、C3及C5位多芳基取代的7-氮杂吲哚衍生物.最近,Kannaboina课题组[16]又报道了一例以5-溴-6-氯-3碘-7-氮杂吲哚为起始原料,通过多次Suzuki-Miyaura偶联反应实现了7-氮杂吲哚C2、C3、C5及C6位的连续芳基化反应.在这些有限的报道中,不仅需要引入氮保护基团,而且大多需要预先在7-氮杂吲哚环引入官能化基团,如卤素、三氟甲磺酰基,进而实现其选择性地芳基化反应.截至目前为止,仅有两例钯催化的直接芳基化报道,均生成C2芳基化产物.因此,本文设计了更为简洁的3位取代的7-氮杂吲哚的芳基化合成路线.从未保护的7-氮杂吲哚出发,通过钯催化下与芳基硼酸的偶联,实现C3位直接芳基化反应.
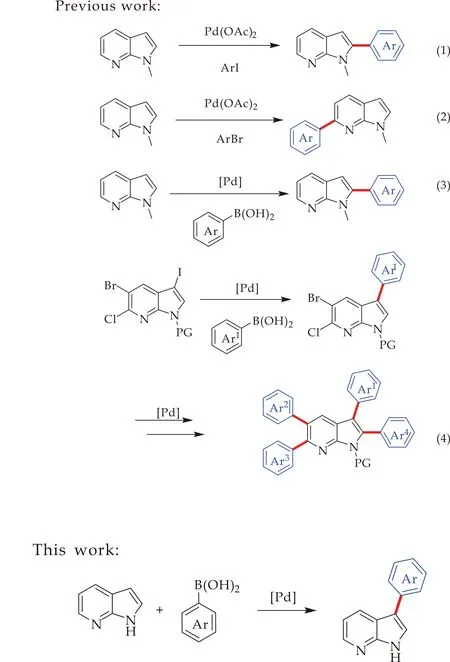
图2 钯催化的7-氮杂吲哚芳基化反应
1 实验部分
1.1 仪器和试剂
1.1.1 主要试剂
苯硼酸,三氟乙酸,甲苯,二甲基乙酰胺,均三甲苯,Cs2CO3(碳酸铯),Cu(OAc)2(醋酸铜),AgOAc(醋酸银),Pd(OAc)2(醋酸钯),Pd(TFA)2(三氟乙酸钯),1,10-phen(1,10-邻菲咯啉).以上试剂均购买自上海萨恩化学技术有限公司;200~300目柱层析用硅胶,青岛海洋化工厂.
1.1.2 主要仪器
AVANCE III 400 MHz型核磁共振仪,德国Bruker公司;Impact HD Q-TOF 型高分辨质谱仪,德国Bruker公司;RE-52A旋转蒸发仪,上海亚荣科技有限公司;MS-H-Pro+型集热式恒温加热磁力搅拌器,大龙兴创实验仪器有限公司.
1.2 实验方法
在15 mL密封管中依次加入7-氮杂吲哚(59.1 mg,0.5 mmol),苯硼酸类化合物(1 mmol),Pd(OAc)2(33.7 mg,0.15 mmol),Cu(OTf)2(361.7 mg,1 mmol),配体(0.2 mmol),CF3CO2H(85.7 mg,0.75 mmol),2.5 mL溶剂,100 ℃下搅拌反应24 h,待反应结束后冷却至室温,用饱和氯化铵水溶液与二氯甲烷萃取三次,得到的有机相用无水硫酸镁干燥,真空旋干,再通过硅胶柱色谱分离得到最终相应的C3位芳基化的7-氮杂吲哚产物.
产品的表征数据如下:
3-(3-氟苯基)-7-氮杂吲哚(3a).无色油状1H NMR(400 MHz,Chloroform-d):δ12.80(s,1H),8.36(s,1H),8.01(d,J=7.7 Hz,1H),7.68(dd,J=23.0,8.7 Hz,2H),7.51(d,J=6.2 Hz,1H),7.21~7.07(m,2H),6.84(s,1H).13C NMR(100 MHz,Chloroform-d):δ163.5,149.4,142.1,138.7,128.3,127.5,123.3,121.8,118.2,117.2,115.9,115.1,112.5 ppm.HRESI-MS:[M+H]+Calcd.for C13H10FN2213.082 3;Found 213.082 5.
3-苯基-7-氮杂吲哚(3b).无色油状(65.1 mg,产率67%).1H NMR(400 MHz,Chloroform-d):δ12.53(s,1H),8.34(d,J=5.7 Hz,1H),8.00(dd,J=7.8,1.3 Hz,1H),7.93(d,J=7.3 Hz,2H),7.55(t,J=7.7 Hz,2H),7.43(t,J=7.4 Hz,1H),7.14(dd,J=7.8,4.8 Hz,1H),6.83(s,1H).13C NMR(100 MHz,Chloroform-d):δ150.1,140.4,129.3,128.7,127.5,123.6,119.2,117.2,116.2,115.9,114.1 ppm.HRESI-MS:[M+H]+Calcd.for C13H10N2195.091 7;Found 195.091 6.
3-(4-氯苯基)-7-氮杂吲哚(3c).黄色油状(68.7 mg,产率60%).1H NMR(400 MHz,Chloroform-d):δ12.42(s,1H),8.34(d,J=4.8 Hz,1H),7.98(dd,J=9.3 Hz,2H),7.76(d,J=8.6 Hz,2H),7.75(s,1H),7.14(s,1H),6.55(s,1H).13C NMR (100 MHz,Chloroform-d):δ149.1,142.4,138.7,129.3,128.9,128.3,123.6,118.2,117.2,115.9,112.1 ppm.HRESI-MS:[M+H]+Calcd.for C13H10ClN2229.052 7;Found 229.052 6.
3-(4-氟苯基)-7-氮杂吲哚(3d).黄色油状(75.3 mg,产率71%).1H NMR(400 MHz,Chloroform-d):δ12.82(s,1H),8.36(d,J=4.7 Hz,1H),7.94(dd,J=8.3 Hz,2H),7.76(d,J= 9.6 Hz,2H),7.71(s,1H),7.14(s,1H),6.85(s,1H).13C NMR (100 MHz,Chloroform-d):δ160.4,149.1,142.4,138.7,129.3,128.9,123.6,118.2,117.2,115.9,112.1 ppm.HRESI-MS:[M+H]+Calcd.for C13H10FN2213.082 3;Found 213.082 3.
3-(4-甲苯基)-7-氮杂吲哚(3e).无色油状(55.2 mg,产率53%).1H NMR(400 MHz,Chloroform-d):δ12.52(s,1H),8.35(d,J=4.7 Hz,1H),7.91(dd,J=8.0 Hz,2H),7.78(d,J=9.4 Hz,2H),7.33(m,1H),7.13(m,1H),6.92(s,1H),2.34(s,3H) ppm.13C NMR(100 MHz,Chloroform-d):δ149.1,142.4,138.7,131.7,128.9,123.6,122.5,118.2,117.2,115.9,114.6,21.3 ppm.HRESI-MS:[M+H]+Calcd.for C13H10FN2209.107 3;Found 209.107 5.
3-(2-甲苯基)-7-氮杂吲哚(3f).无色油状(59.3 mg,产率57%).1H NMR(400 MHz,Chloroform-d):δ12.47(s,1H),8.31(d,J=4.7 Hz,1H),7.92(dd,J=8.5 Hz,1H),7.76(d,J=9.0 Hz,1H),7.40(d,J=9.2 Hz,1H),7.35~7.33(m,2H),7.15(m,1H),6.87(s,1H),2.23(s,3H)ppm.13C NMR(100 MHz,Chloroform-d):δ147.8,141.4,139.2,137.6,131.7,128.9,125.6,124.5,119.2,118.2,117.8,115.9,105.6,27.1 ppm.HRESI-MS:[M+H]+Calcd.for C13H10FN2209.107 3;Found 209.107 4.
3-(4-甲氧基苯基)-7-氮杂吲哚(3g).黄色油状(59.4 mg,产率53%).1H NMR(400 MHz,Chloroform-d):δ12.61(s,1H),8.39(d,J=4.4 Hz,1H),7.94(dd,J=8.3 Hz,2H),7.76(d,J=9.6 Hz,2H),7.33(m,1H),7.15(m,1H),6.87(s,1H),3.87(s,3H) ppm.13C NMR(100 MHz,Chloroform-d):δ147.8,141.4,137.6,131.7,128.9,125.6,124.5,119.2,118.2,115.9,105.6,57.3 ppm.HRESI-MS:[M+H]+Calcd.for C14H13N2O 225.102 2;Found 225.102 2.
3-(4-四氟苯基)-N-苯基-7-氮杂吲哚(3h).无色油状(119.7 mg,产率83%).1H NMR(400 MHz,Chloroform-d):δ8.31 (d,J=4.5 Hz,1H),7.92(dd,J=8.0 Hz,1H),7.56(d,J=7.3 Hz,2H) ,7.50(d,J=7.3 Hz,2H),7.38(t,J=7.4 Hz,1H)7.30(d,J=8.2 Hz,2H),7.20(s,1H),7.15(m,2H),6.87(s,1H),5.54(s,2H)ppm.13C NMR(100 MHz,Chloroform-d):δ162.9,148.0,142.4,137.6,130.7,129.3,128.3,122.9,125.6,121.5,120.7,119.2,116.8,115.9 ppm.HRESI-MS:[M+H]+Calcd.for C19H14FN2289.113 6;Found 289.113 5.
3-(4-四氟苯基)-N-苄基-7-氮杂吲哚(3i).黄色油状(133 mg,产率88%).1H NMR(400 MHz,Chloroform-d):δ8.30(d,J=4.9 Hz,1H),8.00(dd,J=8.0 Hz,1H),7.66(d,J=7.3 Hz,2H),7.58(t,J=7.4 Hz,1H),7.50(d,J=7.3 Hz,2H),7.40(d,J=9.2 Hz,2H),7.20(s,1H),7.15(m,2H),6.87(s,1H)ppm.13C NMR(100 MHz,Chloroform-d):δ162.9,148.0,142.4,137.6,130.7,129.3,128.3,122.9,125.6,121.5,120.7,119.2,116.8,115.9 ppm.HRESI-MS:[M+H]+Calcd.for C20H16FN2303.129 2;Found 303.129 3.
2 结果与讨论
2.1 条件优化
最初,本文选取7-氮杂吲哚(0.5 mmol),3-氟苯硼酸(1.0 mmol)作为7-氮杂吲哚C3位芳基化反应的模板底物,对反应各因素进行了筛选.结果,如图3和表1所示.当溶剂为NMP时,反应产率位35%;溶剂为DMA时,反应产率为43%;溶剂为PhCH3时,产率为40%;溶剂为均三甲苯时,产率为55%.相比之下,溶剂为均三甲苯时反应的产率最高.接下来分别对反应的氧化剂以及配体进行了考察,结果显示,当以PhI(OAc)2或AgOAc为氧化剂时,产率均有下降.以1,1′-双二苯基膦二茂铁(dppf)为配体,产率无明显提高.令人高兴的是,1,10-phen作为配体时,反应的产率最高达到67%.

表1 反应条件筛选
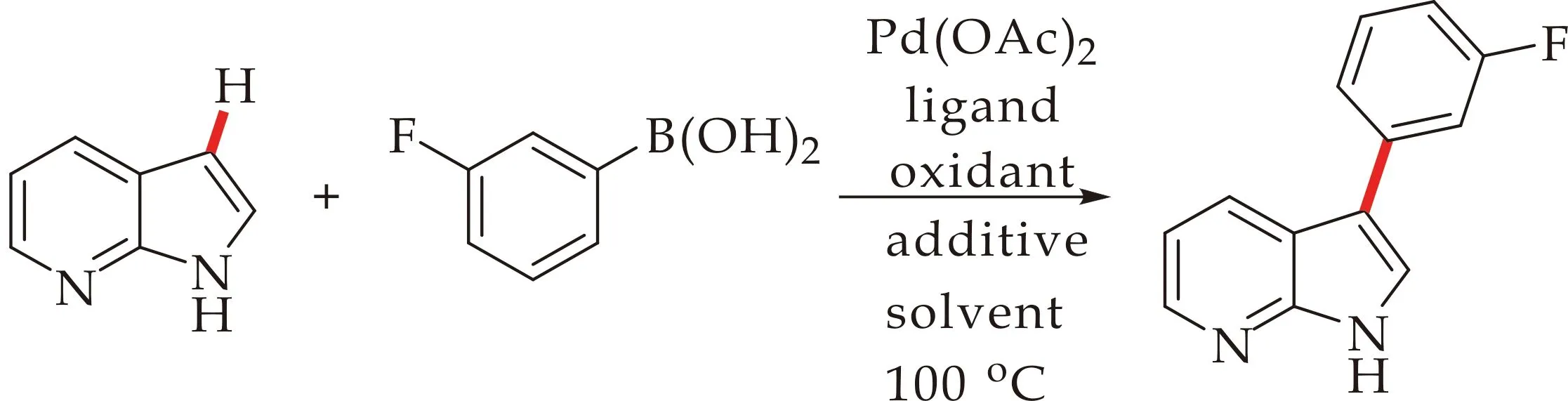
图3 Pd催化的7-氮杂吲哚3位芳基化反应
最后,对反应的催化剂、添加剂和反应温度进行筛选,催化剂为Pd(OAc)2,反应温度为100 ℃时,反应条件最优.考虑到碱性条件对转金属过程有利,本文在反应体系中添加碱,产率却大幅下降,当没有三氟乙酸时,产率也有明显下降,结果表明三氟乙酸对反应产率影响很大,本文考虑可能是由于酸性环境利于提高金属催化剂的亲电性和中间体的活性.因此,根据初步的反应条件探索,本文确定了反应的较优条件为氧化剂为Cu(OTf)2(2.0 equiv),催化剂为Pd(OAc)2(0.3 equiv),配体为1,10-phen(0.4 equiv),添加剂为CF3CO2H(3.0 equiv),反应时间为24 h,反应温度在100 ℃,溶剂为均三甲苯(2.5 mL).
2.2 底物拓展
确定了最优条件后,本文进行了底物拓展(如图4所示).在实验过程中,本文发现底物的电子效应对产率影响较大,而取代基的位置对反应影响可以忽略不计.带有-F、-Cl基团的苯硼酸类化合物与7-氮杂吲哚反应后,产率普遍高于带有甲基、甲氧基的苯硼酸类化合物,可能是有-F、-Cl基团的拉电子效应,更有利于苯硼酸类化合物中C-B键的断裂,进而在金属钯的催化作用下与7-氮杂吲哚中的C3位发生芳基化反应;而7-氮杂吲哚N1位取代的化合物与3-氟苯硼酸反应后,产率也普遍高于7-氮杂吲哚与3-氟苯硼酸反应,可能是7-氮杂吲哚N1位有取代基存在时,可能避免由于N原子与金属配位引起的部分催化剂失活.综上所述,供电子取代基效果明显优于拉电子取代基,因此本文推断反应过程可能会经历亲电金属化步骤,优先在亲核性最强的C3位进行.
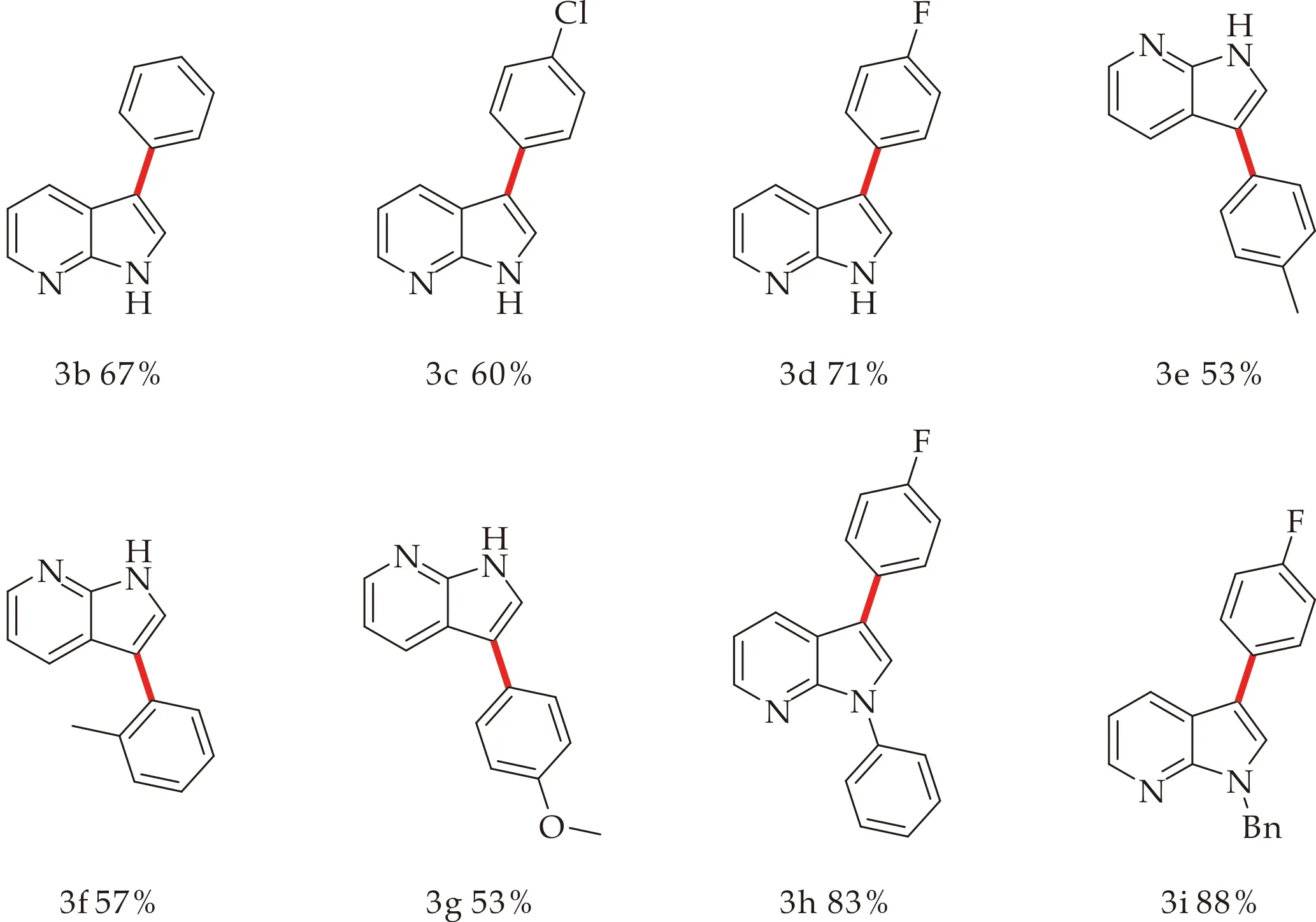
图4 钯催化下7-氮杂吲哚的C3位芳基取代产物的合成
2.3 可能的反应机理
根据前期报道[17],本文推测首先配体与Pd(OAc)2作用,生成Pd配合物,通过脱去一分子AcOH使7-氮杂吲哚的C3-H键活化,并生成活性中间体II,接着与苯硼酸类化合物通过转金属过程发生亲电金属化反应,失去另一份子醋酸根.最后,钯物种III还原消除得到7-氮杂吲哚C3位芳基化产物和零价钯IV.该金属物种在氧化剂的作用下,实现催化剂的再生,构成了催化循环(如图5所示).在本反应中Cu(OTf)2在该催化循环中扮演氧化剂角色,1,10-phen作为配体参与循环,添加剂提供了催化剂再生的醋酸根离子,三者相互作用下,使得反应原料简单,产物易于纯化.
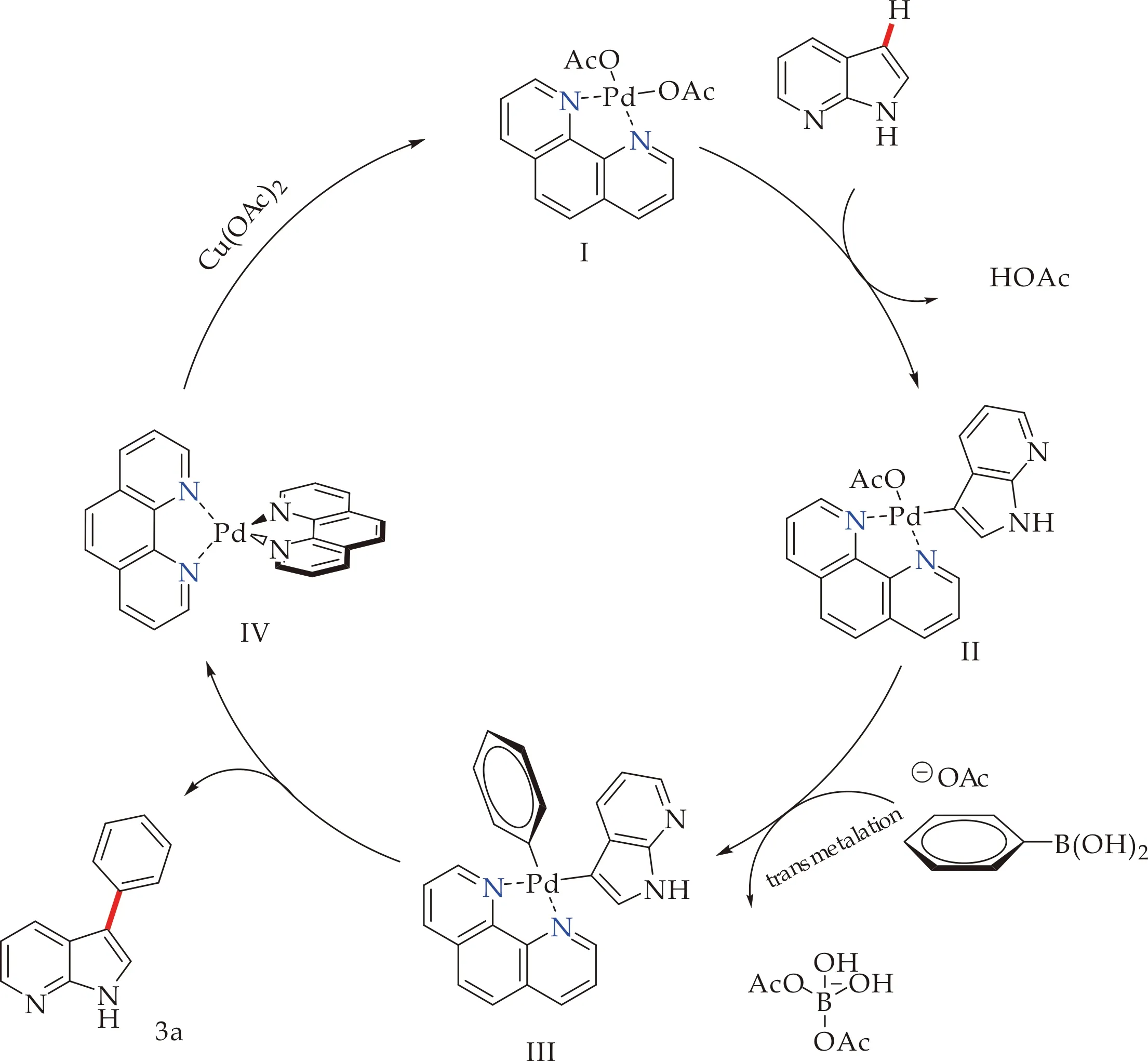
图5 可能的反应机理
3 结论
本文开发了一种钯催化的7-氮杂吲哚C3芳基化的方法,该方法原料廉价易得,合成路线简洁,以53%~88%的收率制备了芳基取代的7-氮杂吲哚的衍生物.初步实验结果符合亲电金属化反应机理.这一钯催化的7-氮杂吲哚3位选择性形成C-C键的反应体系,为7-氮杂吲哚化合物向下游重要生物活性分子转化的方法体系和理论基础.
猜你喜欢
山东第一医科大学(山东省医学科学院)学报(2021年7期)2021-10-13 06:40:42
昆明医科大学学报(2020年12期)2021-01-26 00:44:12
设备管理与维修(2019年9期)2019-09-12 07:44:02
山东化工(2019年11期)2019-06-26 03:26:44
国际呼吸杂志(2019年1期)2019-03-08 03:07:02
合成化学(2015年2期)2016-01-17 09:04:21
大连工业大学学报(2015年4期)2015-12-11 04:06:50
化工进展(2015年6期)2015-11-13 00:27:23
中国塑料(2015年10期)2015-10-14 01:13:13
中国塑料(2015年7期)2015-10-14 01:02:48
