
HDAC抑制剂对食管癌细胞FKBP3的调节作用
2020-09-09 08:25程晓敏丁一楠饶德法刘晓颖
安徽医科大学学报 2020年8期
程晓敏,丁一楠,姬 强,冯 成,饶德法,刘晓颖
中国是食管癌发病率和死亡率较高的国家,亟需更有效的诊断及预后指标。组蛋白去乙酰化酶(histone deacetylases, HDAC)对染色质的结构修饰和基因表达调控有重要作用。正常细胞中,组蛋白乙酰基转移酶(histone acetyltransferase, HAT)与HDAC共同调节组蛋白的乙酰化和去乙酰化,而在恶性肿瘤细胞中这种平衡会被打破,进而影响染色质的结构及基因表达等[1-2]。核心组蛋白N末端的可逆修饰是重构高级染色质结构及控制基因表达的主要表观遗传机制[3]。HDAC抑制剂所导致的开放染色质可上调或抑制基因表达。已知FK506结合蛋白3(FK506 binding protein 3, FKBP3)可通过HDAC2/SP1/p27这一通路调节肿瘤细胞的增殖能力[4],但尚无证据表明HDAC抑制剂可以调节FKBP3。该研究从食管癌细胞入手,探索多种HDAC抑制剂对FKBP3的调节作用,进一步揭示FKBP3/HDACs在肿瘤中的作用。
1 材料与方法
1.1 主要试剂与仪器
1.1.1主要试剂 二甲亚砜(dimethyl sulfoxide, DMSO)购于德国Sigma-Aldrich公司;抑制剂辛二酰苯胺异羟肟酸(suberoylanilide hydroxamic acid, SAHA)、恩替诺特(MS-275)、MC1568和地西他滨(5-AZA)购于上海Selleck公司;胎牛血清购于以色列Biological Industries公司;RPMI-1640和Opti-MEM培养基购自美国GE公司;胰酶细胞消化液、青霉素-链霉素混合液、Western及IP细胞裂解液、苯甲基磺酰氟(PMSF)、Western一抗稀释液购于上海碧云天生物技术有限公司;Lipofectamine RNAi MAX试剂购于美国Thermo-Fisher公司;Total RNA提取试剂盒购于美国Omega公司;NovoScript®Plus All-in-one 1st Strand cDNA 逆转录试剂盒、NovoStart®SYBR qPCR试剂盒 购于江苏近岸蛋白公司;FKBP3抗体购于英国Abcam公司;HDAC1、HDAC2、HDAC3、beta-actin抗体购于武汉Proteintech公司。
1.1.2主要仪器 二氧化碳恒温培养箱(VIOS 160i)、BSC-2级生物安全柜(1300)、微量分光光度计(NanoDrop)、实时荧光定量PCR仪(Quant studio 6 Flex)(美国Thermo-Fischer公司);倒置光学显微镜(TS-100,日本株式会社尼康);高速冷冻离心机(Microfuge 20R,美国Beckman Coulter公司);恒压电泳仪(PowerPacTMBasic,美国BIO-RAD公司);微型垂直电泳槽(VE-180,上海天能科技)。
1.1.3siRNA和qPCR引物 HDAC siRNA和阴性对照siRNA合成于上海吉玛生物科技有限公司,序列见表1。qPCR引物合成于通用生物系统(安徽)有限公司,序列见表2。
1.1.4细胞 食管癌细胞系ECA109和KYSE30为本实验室保存。

表2 qPCR引物序列
1.2 实验方法
1.2.1细胞培养和加药实验 ECA109和KYSE30细胞系使用含10%胎牛血清的RPMI-1640培养基,置于37 ℃、5% CO2、95%湿度的培养箱中分别培养;培养的细胞每1~2 d换液1次,细胞密度大于90%时进行传代,使用胰酶对细胞进行消化,以培养基终止消化反应并计数。加药实验时,按照3×105个/孔的数量接种于12孔板,待细胞贴壁生长稳定后,在培养基中加入药物:SAHA(终浓度为4 μmol/L,下同)、MS-275(5 μmol/L)、MC1568(5 μmol/L)、5-AZA(5 μmol/L);设置未处理和阴性对照(加入1 ∶1 000 的DMSO)组;连续培养48 h后,胰酶消化并收集细胞。
1.2.2siRNA knockdown实验 转染前1 d,将待转染细胞以1.5×105个/孔的数量接种于6孔板。次日观察贴壁后,使用Opti-MEM以50 nmol/L的终浓度稀释siRNA储液;再以1 ∶1的比例稀释Lipofectamine RNAi MAX试剂,将二者混匀,静置15 min;弃尽培养皿中的培养液,加入上述混合转染液继续培养;转染6 h后更换新鲜培养液;转染48 h后,胰酶消化并收集细胞以进行后续实验。
1.2.3逆转录和qPCR 使用Total RNA 提取试剂盒对收集的细胞进行总RNA提取,NanoDrop 2000分光光度计测定RNA浓度;按试剂盒要求进行逆转录,获得的cDNA保存于-80 ℃;按试剂盒要求进行实时荧光定量PCR扩增,并以GAPDH为内参,通过2-ΔΔCt法分析相对基因表达,计算各组mRNA的相对含量。
1.2.4Western blot 加入适量Western细胞裂解液裂解收集的细胞,冰浴30 min;4 ℃、14 000 r/min离心20 min;收集上清液,Bradford法进行总蛋白浓度测定;各取30 μg总蛋白样品,沸水浴5 min变性,SDS-PAGE电泳分离目的蛋白,转移蛋白于PVDF膜;5%脱脂牛奶于室温封闭1.5 h,TBST室温洗膜3次,每次10 min;一抗孵育4 ℃过夜,TBST洗膜3次;室温孵育二抗1.5 h,TBST洗膜3次;ECL显影液显影。使用ImageJ软件(美国国立卫生研究院)进行条带的光密度分析,并以β-actin为内参计算FKBP3的相对表达。
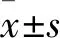
2 结果
2.1 HDAC抑制剂抑制食管癌细胞FKBP3的表达对培养的ECA109和KYSE30细胞进行HDAC抑制剂加药处理,Western blot结果显示SAHA和MS-275处理48 h后,FKBP3的蛋白含量有所降低,而MC1568、5-AZA和DMSO处理后的细胞,其FKBP3的含量没有明显变化。使用ImageJ对条带的灰度值进行测定,以β-actin作为内参进行半定量分析,对3次重复实验的结果进行汇总,同时以DMSO组的数据为基准,制作加药后FKBP3蛋白相对含量的直方图(图1)。依据图1的数据,HDAC抑制剂SAHA及MS-275处理48 h对ECA109细胞及KYSE30细胞中的FKBP3蛋白表达有一定的抑制作用,但统计分析该结果差异无统计学意义(P>0.05)。
经qPCR实验结果显示FKBP3的mRNA水平同样被SAHA和MS-275所抑制(图2)。在ECA109细胞中,经2-ΔΔCt法分析后,FKBP3的mRNA含量为DMSO:(0.039±0.003);SAHA:(0.020±0.001);MS-275:(0.017±0.005)。在KYSE30细胞中为DMSO:(0.022±0.001);SAHA:(0.008±0.001);MS-275:(0.009±0.001)。这说明SAHA和MS-275对于FKBP3的抑制作用起始于mRNA水平。鉴于SAHA是广谱的HDAC抑制剂,而MS-275特异性抑制HDAC1和HDAC3,提示HDAC对FKBP3表达的影响,可能主要是通过HDAC1发挥作用的。
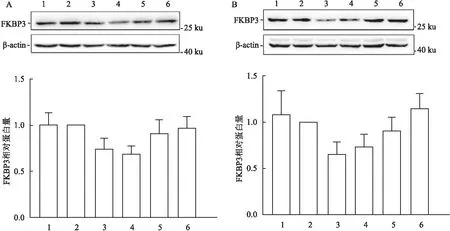
图1 Western blot法检测HDAC抑制剂处理后食管癌细胞FKBP3表达

图2 qPCR检测HDAC抑制剂对食管癌细胞FKBP3 mRNA水平的影响
2.2 HDAC1和HDAC2调节FKBP3的表达为了进一步确定FKBP3被HDAC调控的具体机制,在ECA109细胞中进行了HDAC1、HDAC2、HDAC3的siRNA敲低实验。经qPCR结果显示,转染48 h后,对比阴性对照组(siNC),3种siRNA敲低组的对应HDAC基因的mRNA的相对含量均减少60%以上(图3)。Western blot结果显示,HDAC1和HDAC2敲低的细胞中,FKBP3的蛋白表达受到一定程度的抑制,而HDAC3的敲低对FKBP3没有明显影响(图4)。该结果表明,HDACs对FKBP3的调控很可能是通过HDAC1或HDAC2实现。
2.3 对FKBP3和HDAC相互作用的预测在STRING数据库中检索人类FKBP3相互作用蛋白(图5)。图5中显示与FKBP3相互作用置信度最高的10个蛋白,HDAC2和HDAC1蛋白分别位于FKBP3相互作用列表的第1、2位,得分分别为0.958和0.954分(分数区间为0~1分,1分代表相互作用程度最高);FKBP3和HDAC2、HDAC1的相互作用,均获得了直接实验及文献资料库的支持。并且,根据NCI-Nature Pathways Interaction Database数据库,FKBP3、HDAC1、HDAC2 这3个蛋白可以作为一个复合体,共同发挥生物学功能。其他诸如CSNK2A1、YY1、哺乳动物雷帕霉素靶标蛋白(mammalian target of rapamycin, mTOR)等蛋白也存在于该相互作用网络中。

图3 qPCR检测ECA109细胞中HDAC siRNA的敲低效果

图4 ECA109细胞中HDAC siRNA敲低后的
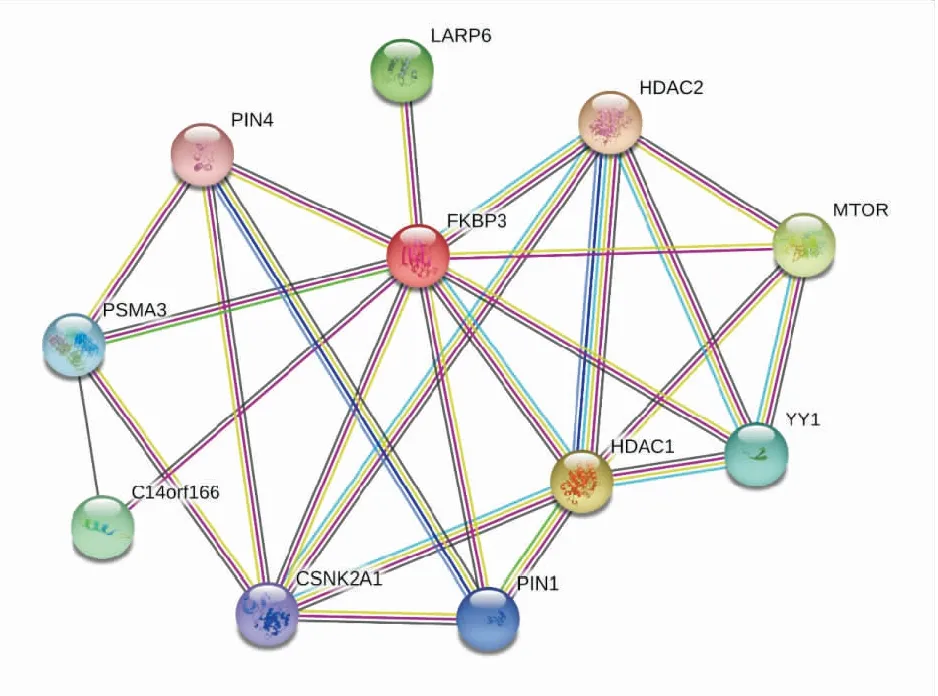
图5 STRING数据库中FKBP3的蛋白相互作用网络
3 讨论
组蛋白乙酰化是基因表达的重要决定因素。乙酰化通常与转录活化有关,而去乙酰化的组蛋白通常与转录抑制有关。HDAC是基因表达的关键调节因子,可以从核心组蛋白中去除乙酰基。已有研究表明在结肠癌、乳腺癌和肺癌等多种恶性肿瘤中存在HDAC的异常表达[5]。
近年来,随着研究的深入和临床试验的推进,HDAC抑制剂已被广泛应用于恶性肿瘤的治疗中[6],如美国FDA于2006年批准SAHA用于顽固性T细胞淋巴瘤,于2014年批准PXD101用于周围性T细胞淋巴瘤。本研究结果表明,SAHA和MS-275不仅可以调节HDAC的水平,还可以调节HDAC相互作用蛋白FKBP3,并且SAHA和MS-275对于FKBP3的调节作用是在mRNA水平发挥作用的。进一步研究显示,FKBP3蛋白受HDAC调节主要来自于HDAC1和HDAC2,而非HDAC3。本研究中,经SAHA和MS-275处理后的食管癌细胞,对FKBP3的mRNA抑制效应比蛋白水平更显著,推测可能是蛋白水平的变化存在滞后性,以及FKBP3的翻译后修饰增强了蛋白的稳定性。
FKBP3是PPIase超家族的一员,其蛋白的C端是FKBP家族共有的PPIase结构域,并且也是FK506结合结构域,与免疫抑制剂FK506、雷帕霉素(Rapamycin)均可结合。在哺乳动物组织中发现的18种FKBPs中,FKBP3和Rapamycin的结合能力仅次于FKBP12,已证明FKBP3的FRB区域可以通过Rapamycin在活细胞中产生物理结合[7],这为FKBP3结合并影响mTOR提供了依据。mTOR作为磷脂酰肌醇3激酶(phosphatidylinositol 3 kinase, PI3K)相关激酶,参与许多细胞增殖和细胞周期相关的重要调节功能,包括在PI3K/Akt通路的下游发挥作用并被磷酸化,进而调节4E-BP1和70S核糖体蛋白6激酶(p70 S6K)通路。mTOR在肿瘤组织中的表达也明显高于正常组织,是研究肿瘤发生和开发新型抗肿瘤药物的重点靶点。乙酰化是表观遗传修饰的重要机制,组蛋白的乙酰化一般伴随基因的活化,有报道称Rapamycin抑制mTOR后可以使Esa1酶从核糖体蛋白基因的启动子释放,导致组蛋白H4去乙酰化[8]。mTOR和HDACs的关系密切,PI3K/AKT/mTOR通路调节HDAC3 S424的磷酸化,从而促进HDAC3和磷酸甘油酸激酶1(phosphoglycerate kinase 1, PGK1)相互作用及PGK1 K220去乙酰化[9]。Rapamycin和MS-275联用也被报道在多种癌症治疗中有效,可显著下调MYC和E2F1的表达,进而抑制肿瘤细胞生长[10]。基于FKBP3对Rapamycin有较强的结合能力,提示FKBP3在细胞周期调控中发挥作用,可能为潜在的肿瘤药物靶点。
STRING数据库整合了基因组背景、高通量测序、共表达、已发表文本挖掘和其他数据库内容[11]。该数据库的结果进一步预测了HDAC1、HDAC2和FKBP3之间的相互作用,并且提示这三者可能共同组成复合物发挥作用。综上,本研究表明,HDAC抑制剂可能在mRNA水平调节FKBP3的表达,而且很可能通过HDAC1、HDAC2实现的,具体分子机制还需进一步研究。
猜你喜欢
中国生物化学与分子生物学报(2022年8期)2022-09-08
科教新报(2022年10期)2022-03-27
家庭医药(2022年3期)2022-03-24
复旦学报(自然科学版)(2021年5期)2021-11-17
天津医科大学学报(2021年4期)2021-08-21
现代仪器与医疗(2021年2期)2021-07-21
现代临床医学(2021年2期)2021-03-29
医药前沿(2020年29期)2020-12-04
山西医科大学学报(2020年8期)2020-09-16
腹腔镜外科杂志(2016年9期)2016-06-01
