
藏药材诃子中3种α-葡萄糖苷酶抑制剂的分离与鉴定
2020-08-31 03:45:44马家麟李玉林
天然产物研究与开发 2020年8期
马家麟,陈 涛,谭 亮,王 环,李玉林
中国科学院西北高原生物研究所 中国科学院三江源国家公园研究院,西宁 810008
α-葡萄糖苷酶是与糖尿病密切相关的酶之一,对碳水化合物的代谢至关重要,α-葡萄糖苷酶抑制剂能通过竞争性抑制酶的活性从而延缓碳水化合物的吸收,降低餐后和空腹的血糖水平[1-3],有效控制病情发展。目前市场上使用的α-葡萄糖苷酶抑制剂主要分为人工合成和天然来源两大类,人工合成的α-葡萄糖苷酶抑制剂如阿卡波糖等有腹泻、腹胀痛、肝肾损伤等副作用,严重阻碍了其广泛应用[4]。而天然来源的α-葡萄糖苷酶抑制剂具有疗效稳定、安全、毒副作用小等优势,是现代医药领域的研究热点[5,6]。
诃子为使君子科植物诃子(TerminaliachebulaRetz.)或绒毛诃子(T.chebulaRetz.var.tomentellaKurt)的干燥成熟果实,规肺,大肠经[7],具有涩肠止泻、敛肺止咳、降火利咽等功效[8]。现代医学研究表明,诃子具有抗氧化、抗病毒、抗肿瘤、强心等药理作用[9],而且,诃子所含的酚酸类物质在体外和体内均表现出良好的抗糖尿病活性[10,11],这表明诃子可能含有潜在的α-葡萄糖苷酶抑制剂。
目前关于诃子提取物中的抗糖尿病活性的成分的文献报道甚少,因此,亟需深入研究建立高效的检测技术,从诃子中筛选分离天然α-葡萄糖苷酶抑制剂。
1 材料与仪器
1.1 材料
诃子药材采购自青海省西宁市八一路藏药材市场,经中国科学院西北高原生物研究所孙菁研究员鉴定为诃子(TerminaliachebulaRetz.)干燥果实。
1.2 试剂
酵母中的α-葡萄糖苷酶(美国西格玛-奥德里奇公司);阿卡波糖(德国拜耳公司);4-硝基酚-α-D-吡喃葡萄糖苷(pNPG,美国西格玛-奥德里奇公司);D101、HPD-600、S-8、X-5、AB-8、D3520大孔吸附树脂(沧州宝恩吸附材料科技有限公司);用于高效液相色谱分析的甲醇为色谱纯;用于大孔吸附树脂分离的乙醇和半制备高效液相色谱的甲醇为分析纯;二甲基亚砜及方法中各化学试剂均为分析纯;实验用水为去离子水。
1.3 仪器
LC-8A半制备高效液相色谱(SPD-20A紫外可见光检测器,日本岛津公司);Dubhe C18柱(250 mm× 10 mm,10 μm);Aglient 1260系列高效液相色谱仪(G1315D紫外可见光二极管阵列检测器,美国Aglient公司);Dikma Platisil ODS-C18分析柱(250 mm× 4.6 mm,5 μm);安亭TGL-16C系列离心机(上海安亭科学仪器厂);10 kDa超滤离心管(德国Merck公司);Varian Mercury-400B核磁共振波谱仪(美国Varian公司);Varian Cary 300紫外可见分光光度计(美国Varian公司)。
2 实验方法
2.1 诃子提取物中α-葡萄糖苷酶抑制剂的筛选
2.1.1 样品前处理
清除诃子药材表面灰尘杂质,称取1.0 kg粉碎过40目筛的诃子粉末,料液比1∶15(V/V)加入75%乙醇加热回流提取1 h,过滤,药渣再加入75%乙醇15 L,反复提取3次,合并滤液,减压浓缩蒸干,得到诃子粗提物约210 g,冷藏备用。
2.1.2 超滤技术筛选α-葡萄糖苷酶抑制剂
精密称取50.0 mg诃子粗提物至50 mL容量瓶中,加水溶解后定容至刻度,摇匀。用0.45 μm滤膜过滤,即得。分别准确移取250 μL供试品溶液和50 μL 0.5 U/mLα-葡萄糖苷酶至10 mmol/L醋酸铵缓冲液(pH6.8)中,37 ℃反应30 min。然后移取100 μL混合溶液至10 kDa的超滤离心管中,在10 000 rpm下离心15 min,用10 mmol/L醋酸铵缓冲液离心清洗滤液三次,每次100 μL,以除去游离成分,未被滤过的成分用于液相色谱分析。
2.1.3 色谱分析条件
色谱柱:Dikma Platisil ODS-C18(250 mm× 4.6 mm,5 μm);流动相:0.1%乙酸-水溶液(A相),甲醇(B相),梯度洗脱(0~30 min,30%→70%B);流速:1.0 mL/min;检测波长:254 nm;柱温:25 ℃;进样量:10 μL。
2.2 大孔吸附树脂富集诃子中α-葡萄糖苷酶抑制剂
2.2.1 大孔吸附树脂的选择
考察6种大孔吸附树脂(D101、HPD-600、S-8、X-5、AB-8和D3520)的对目标化合物的富集效果。将大孔吸附树脂用95%乙醇浸泡24 h,再用大量去离子水冲洗至流出液无醇味。然后依次用4%盐酸溶液、去离子水和4%氢氧化钠溶液各清洗一次,最后用去离子水冲洗至流出液呈中性,处理好的树脂晾干备用。
2.2.2 静态吸附实验
分别称取预处理后的6种大孔吸附树脂2.0 g,精密称定,置于50 mL具塞锥形瓶中,各加入诃子粗提物样品溶液(20 mg/mL)25 mL,密封后于室温条件下以120 rpm转速振摇12 h使吸附完全,过滤,定容至25 mL,测定反应前后目标化合物的峰面积,按式(1)计算吸附率。过滤后,干燥固体树脂,收集备用。
Q=(P1-P2)/P1×100%
(1)
其中,Q为吸附率;P1为吸附前样品溶液中目标化合物峰面积;P2为吸附后溶液中目标化合物峰面积。
2.2.3 静态解吸附试验
将静态吸附后得到的6种树脂分别置于50 mL具塞锥形瓶中,各加95%乙醇溶液25 mL,密封后于室温条件下以120 rpm转速振摇12 h使解吸附完全,过滤,定容至25 mL,测定溶液中目标化合物的峰面积,按式(2)计算解析率。
D=P3/P1×100%
(2)
其中,D为解析率;P1为解吸附前样品溶液中目标化合物峰面积;P3为解吸附后溶液中目标化合物峰面积。
2.3 分离条件优化
2.3.1 洗脱剂浓度考察
取用“2.2”中选定的大孔树脂,湿法装柱,测定柱体积为200 mL,注入100 mL诃子粗提物样品溶液,静置24 h,分别用10%、20%、30%、40%、50%、60%、70%、80%和90%乙醇溶液冲洗大孔树脂柱,每个浓度均冲洗5倍柱体积,收集洗脱液,用高效液相色谱法测定每个洗脱组分中目标化合物的峰面积。
2.3.2 洗脱体积考察
取用“2.2”中选定的大孔树脂,湿法装柱,测定柱体积为200 mL,注入100 mL诃子粗提物样品溶液,静置24 h。首先用30%乙醇洗脱大孔树脂柱,除去非目标组分,采用高效液相色谱法测定每个洗脱体积中目标化合物的峰面积,绘制洗脱曲线,根据洗脱曲线确定洗脱体积。然后,采用50%乙醇溶液洗脱大孔树脂柱,富集目标化合物,用高效液相色谱法测定每个洗脱体积中目标化合物的峰面积,绘制洗脱曲线,根据洗脱曲线确定洗脱体积。
2.4 目标化合物的纯化与鉴定
2.4.1 半制备高效液相色谱分析条件
色谱柱:Dubhe C18柱(250 × 10 mm,10 μm);0.1%乙酸(A相),甲醇(B相);梯度洗脱(0~50 min,20%→70%B);流速:4.5 mL/min;检测波长:254 nm;进样量:5 mL。
2.4.2 目标化合物结构鉴定
将半制备高效液相色谱分离得到的3个组分用二甲基亚砜溶解,以TMS为内标物,根据核磁共振1H NMR 和13C NMR波谱数据鉴定3个组分结构。
2.5 诃子中目标化合物的α-葡萄糖苷酶体外抑制活性检测
活性检测反应体系配制如下[12,13]:取0.1 mol/L磷酸缓冲液(pH6.8)30 μL,分别加入二甲基亚砜5 μL和0.1 U/mLα-葡萄糖苷酶10 μL,37 ℃反应10 min,加入1.0 mmol/L pNPG 10 μL,37 ℃反应15 min,再加入0.1 mol/L Na2CO3溶液45 μL终止反应,混匀后,于405 nm波长下测定吸光度A值,每个化合物3次平行实验。以对α-葡萄糖苷酶有显著抑制作用的阿卡波糖作为阳性对照,按照I=[1-(A样品组-A样品空白组)/(A阴性组-A空白组)]×100%,计算抑制率I,并用Origin软件计算IC50值。
3 结果与分析
3.1 诃子提取物中α-葡萄糖苷酶抑制剂筛选
液相检测结果如图1所示,a为诃子提取物溶液(对照组);b为与α-葡萄糖苷酶超滤亲和后的混合液(试验组)。由图可见,实验组混合液中主要含有的化合物为1~3,说明这3个化合物与α-葡萄糖苷酶发生特异性结合,未透过超滤膜,因此,以上3个化合物被认为是α-葡萄糖苷酶抑制剂,而其他化合物没有α-葡萄糖苷酶抑制活性。以化合物1~3作为目标化合物,对其进行分离制备。
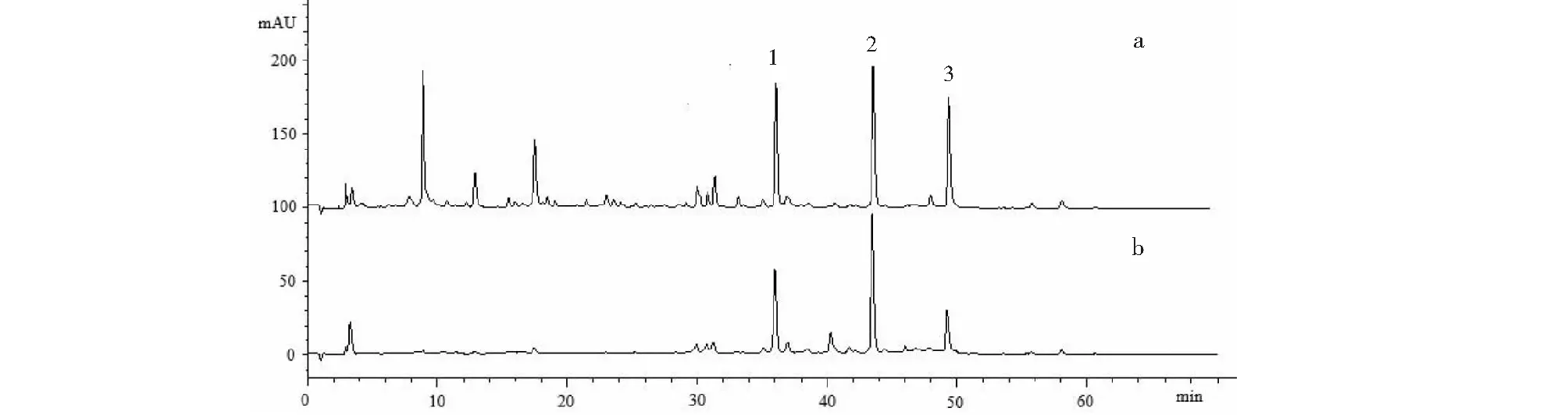
图1 对照组(a)和试验组(b)液相色谱图Fig.1 HPLC chromatograms of the control sample (a) and the experimental sample (b)
3.2 大孔吸附树脂富集诃子中α-葡萄糖苷酶抑制剂
3.2.1 大孔吸附树脂的选择
由图2可知,在考察的D101、HPD-600、S-8、X-5、AB-8和D3520 6种大孔吸附树脂中,AB-8和X-5树脂的吸附性能优于其它树脂。由图3可知,AB-8和D101树脂的解吸率高于其他树脂。因此,AB-8大孔吸附树脂对于目标化合物的吸附和解吸附能力均有良好表现,选择AB-8树脂进行后续的分离。

图2 6种树脂对化合物1~3的吸附率考察Fig.2 Adsorption ratio of compounds 1-3 on different macroporous resin
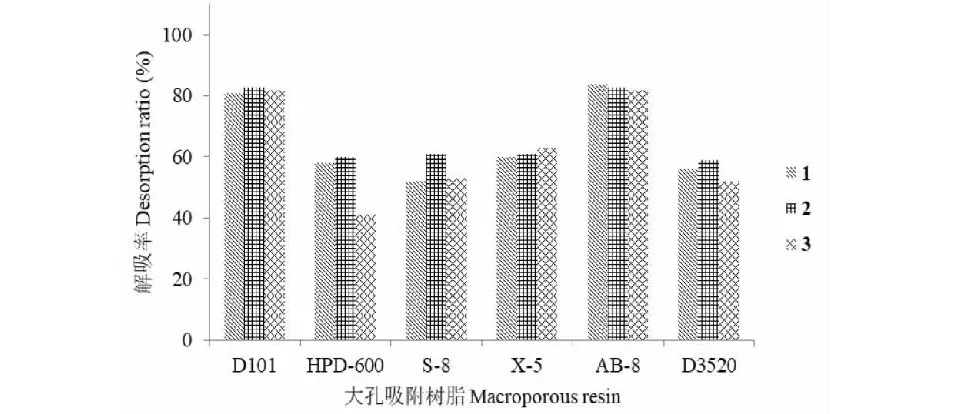
图3 6种树脂对化合物1~3的解析率考察Fig.3 Desorption ratio of compounds 1-3 on different macroporous resin
3.2.2 分离条件优化
由表1可知,目标化合物主要分布在40%乙醇中。30%乙醇中的化合物1峰面积占比为12.92%,50%乙醇中的化合物2峰面积占比为10.41%。因此,有必要选择合适体积的30%乙醇去除杂质,再用40%乙醇洗脱目标化合物。
用30%乙醇洗脱目标化合物时,洗脱曲线如图4所示。结果表明:当洗脱体积从4 BV增加到5 BV时,洗脱液中化合物1的峰面积明显增高,因此,选择使用4 BV 30%乙醇去除非目标化合物。然后,用40%乙醇溶液洗脱目标物,洗脱曲线见图5。结果表明:当洗脱体积增加到8 BV时,洗脱液中化合物3的峰面积变化趋近于零。因此,选择7 BV 40%乙醇溶液洗脱目标物。
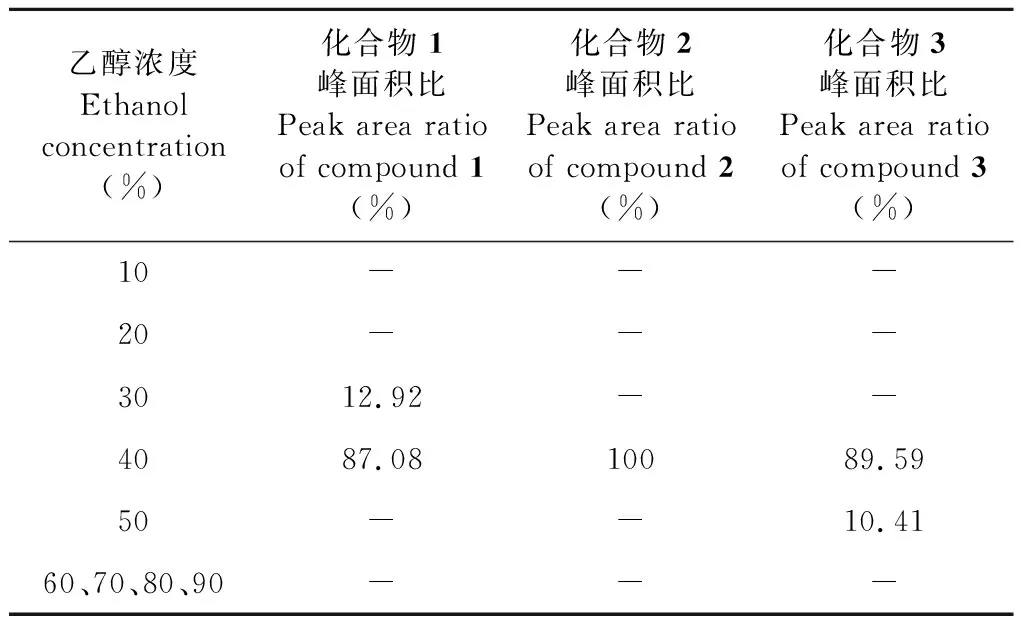
表1 3种组分在不同浓度洗涤液中的含量
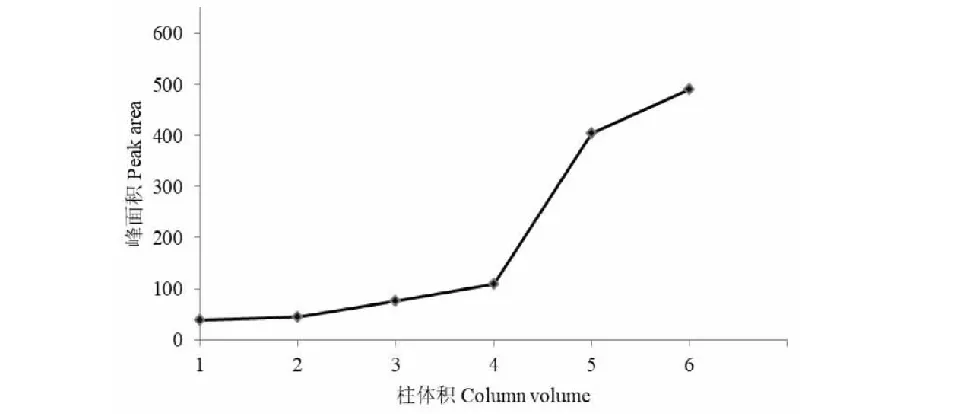
图4 不同柱体积的30%乙醇对化合物1的洗脱曲线Fig.4 The elution curve of compound 1 on AB-8 column

图5 不同柱体积的50%乙醇对化合物3的洗脱曲线Fig.5 The elution curve of compound 3 on AB-8 column
选择AB-8树脂对诃子粗提物中3种α-葡萄糖苷酶抑制剂进行富集,采用优化后的条件,从113 g诃子粗提物中得到34.6 g淡黄色粉末状提取物,得率为30.6%。诃子提取物中目标化合物的峰面积比分别由15.8%、19.1%和11.2%,提高至29.6%、40.3%和21.9%,提取率平均增加了2倍,化合物纯度显著提高,为后续目标物纯化工作奠定了基础。
3.3 目标化合物的纯化与鉴定
3.3.1 目标化合物的纯化
由半制备高效液相色谱仪对大孔树脂富集产物进行分离,并采集各目标组分,减压蒸干,从100 mg样品中得到化合物1(27 mg)、化合物2(35 mg)和化合物3(22 mg),3个化合物纯度分别为96.6%、98.1%和96.4%。
3.3.2 目标化合物结构鉴定
化合物1白色粉末;易溶于甲醇,微溶于水,不溶于乙酸乙酯、苯、丙酮、氯仿;1H NMR(400 MHz,CD3OD)δ:6.49(1H,s,H-1),5.39(1H,brs,H-2),5.82(1H,brs,H-3),5.23(1H,d,J=3.0 Hz,H-4),4.83(1H,t,J=17.4 Hz,H-5),4.37(1H,dd,J=10.2,7.8 Hz,H-6a),4.90(1H,t,J=20.3 Hz,H-6b);4.79(1H,d,J=7.2 Hz,H-2′),5.04(1H,dd,J=7.3,1.2 Hz,H-3′),3.80(1H,m,H-4′),2.15(1H,m,H-5′),7.48(1H,s,H-2″);7.07(2H,s);6.84(2H,s);6.63(2H,s);13C NMR(100 MHz,CD3OD)δ:91.1(C-1),69.7(C-2),61.0(C-3),65.6(C-4),72.9(C-5),63.3(C-6);169.4(C-1′),65.4(C-2′),40.3(C-3′),38.6(C-4′),29.2(C-5′),173.6(C-6′),173.0(C-7′),114.8(C-1″),117.6(C-2″),116.2(C-3″),146.0(C-4″),139.0(C-5″),140.0(C-6″),165.0(C-7″);118.7(C-1″′),109.5(C-2″′,6″′),145.1(C-3″′,5″′),139.5(C-4″′),164.9(C-7″′);114.6(C-1),124.2(C-2),106.8(C-3),144.8(C-4),136.2(C-5),143.9(C-6),168.7(C-7);116.3(C-1′),123.1(C-2′),109.1(C-3′),144.2(C-4′),137.3(C-5′),144.1(C-6′),166.1(C-7′)。以上数据与文献报道一致[14],故鉴定化合物1为诃黎勒酸(见图6-1)。
化合物2白色粉末,易溶于甲醇,微溶于水,不溶于乙酸乙酯、苯、丙酮,氯仿。1H NMR(400 MHz,CD3OD)δ:6.49(1H,d,J=2.9 Hz,H-1),5.42(1H,brs,H-2),6.25(1H,brs,H-3),5.04(1H,d,J=3.0 Hz,H-4),4.72(1H,dddd,J=7.8,6.6,1.1,1.0 Hz,H-5),4.61(1H,dd,J=10.9,6.6 Hz,H-6a),4.79(1H,dd,J=11.0,7.8 Hz,H-6b);4.80(1H,d,J=7.1 Hz,H-2′),5.06(1H,dd,J=7.2,1.3 Hz,H-3′),3.87(1H,ddd,J=10.7,4.2,1.4 Hz,H-4′),2.22(1H,m,H-5′a),2.24(1H,m,H-5′b),7.51(1H,s,H-2″);7.11(2H,s);7.16(2H,s);6.98(2H,s);13C NMR(100 MHz,CD3OD)δ:91.6(C-1),70.8(C-2),61.7(C-3),68.1(C-4),74.9(C-5),63.8(C-6);169.4(C-1′),65.8(C-2′),40.4(C-3′),39.1(C-4′),30.1(C-5′),174.6(C-6′),173.3(C-7′),117.6(C-1″),116.2(C-2″),146.0(C-3″),139.2(C-4″),140.1(C-5″),115.0(C-6″),164.7(C-7″);118.6(C-1″′),109.3(C-2″′,6″′),145.2(C-3″′,5″′),139.4(C-4″′),164.9(C-7″′); 118.5(C-1″′),109.4(C-2″′,6″′),145.3(C-3″′,5″′),139.4(C-4″′),164.8(C-7″′);119.3(C-1″′),110.4(C-2″′,6″′),145.0(C-3″′,5″′),138.7(C-4″′),166.5(C-7″′)。数据与文献报道基本一致[14],故鉴定化合物2为诃子酸(见图6-2)。
化合物3黄色粉末,易溶于甲醇,微溶于水。1H NMR(400 MHz,DMSO-d6)δ:7.45(2H,s,H-5/H-5′),4.80(2H,br,H-6/H-6′);13C NMR(100 MHz,DMSO-d6)δ:107.7(C-5),110.3(C-6),112.3(C-1),136.4(C-2);139.5(C-3),148.1(C-4),159.1(C-7)。数据与文献报道基本一致[15],故鉴定化合物3为鞣花酸(见图6-3)。
3.4 诃子中目标化合物的α-葡萄糖苷酶抑制活性检测
由表2可知,与阳性对照阿卡波糖比较,从诃子中分离得到的3种α-葡萄糖苷酶抑制剂均表现出良好的抑制活性,其中,诃黎勒酸和诃子酸抑制作用最强,其IC50分别为39.2±0.7,35.8±0.4 mg/L,抑制率分别达到95.9%、98.3%。

图6 化合物1~3的化学结构Fig.6 The chemical structures of compound 1-3

表2 α-葡萄糖苷酶抑制活性
4 结论与讨论
经α-葡萄糖苷酶反应后,药材提取物中具有亲和活性的化合物配体与α-葡萄糖苷酶结合成大分子量的混合物,其他没有亲和力的化合物则处于小分子游离状态。使混合溶液通过超滤膜时,与α-葡萄糖苷酶结合的配体化合物将无法通过滤膜,而游离态的化合物被滤过,未透过滤膜的物质经高效液相色谱分析,对比提取物液相色谱图,观察化合物峰面积变化,即可以达到快速筛选α-葡萄糖苷酶抑制剂的目的。
诃子是最常用的藏药,在藏药学经典著作《晶珠本草》里,诃子被称为“藏药之王”,其活性成分主要是鞣质、酚酸、三萜类、黄酮类物质[10]。本文以诃子为研究目标,首次采用超滤高效液相色谱法、大孔树脂法、半制备高效液相色谱法结合核磁共振色谱法快速筛选、分离和鉴定出诃子中3种具有α-葡萄糖苷酶抑制活性的成分,最终鉴定这3种化合物分别为诃黎勒酸、诃子酸和鞣花酸。活性检测结果显示,这3种酚酸类成分能与α-葡萄糖苷酶高效结合,显示该化合物具有良好的α-葡萄糖苷酶抑制活性,表明诃子中存在具有糖尿病治疗药物开发潜力的成分,为开发基于诃子的降糖药物提供了科学参考依据。
猜你喜欢
河北农业(2022年8期)2022-09-28 06:59:32
陶瓷学报(2020年6期)2021-01-26 00:38:14
中国林副特产(2019年6期)2019-01-11 02:33:06
制造技术与机床(2018年10期)2018-10-13 06:36:56
中成药(2018年5期)2018-06-06 03:11:58
中成药(2017年8期)2017-11-22 03:19:32
西藏科技(2016年9期)2016-09-26 12:21:39
山东医药(2015年14期)2015-04-04 14:00:07
天然产物研究与开发(2014年3期)2014-04-27 14:15:35
茶叶通讯(2014年4期)2014-02-27 07:55:49
