
基于生物信息学方法筛选与肝细胞癌发生和预后相关的潜在关键基因
2020-08-10 09:26石华盛辛洋韩冰吴力群
精准医学杂志 2020年4期
石华盛 辛洋 韩冰 吴力群
(青岛大学附属医院,山东 青岛 266003 1 肝脏病中心; 2 肝胆胰外科)
肝细胞癌(HCC)仍是目前全球范围内最常见的恶性肿瘤之一,每年新发病例和死亡病例均超过80万例,而我国患者的占比超过50%[1]。尽管近年来对HCC的发生和发展已经进行了深入的研究,但HCC发生的确切分子机制仍不是很清楚。同时由于治疗策略的局限性,全世界的HCC病死率依旧很高,因此,迫切需要了解HCC发生发展的确切机制。近年来,随着基因检测技术的不断发展和逐渐成熟,微阵列和高通量测序技术在探索肿瘤诊断、治疗和预后相关的生物标志物方面发挥越来越大的作用[2],如影响胃癌预后的关键基因就首先通过生物信息学分析技术预测并最后证实的[3]。因此通过对HCC的基因表达谱进行生物信息学分析可有效帮助了解肿瘤形成的分子机制,甚至可以找到潜在的治疗新靶点。本研究首先利用公共数据库中的RNA测序数据,筛选人类HCC组织和正常肝组织样本中差异表达的基因,再对这些差异表达的基因进行功能富集分析,探究其主要参与调控的生物学功能,并通过PPI网络和生存分析,挖掘HCC发病和预后的潜在关键基因。
1 资料与方法
1.1 通过GEO和TCGA数据库筛选HCC组织与正常肝组织差异表达基因
首先通过Gene Expression Omnibus(GEO,https://www.ncbi.nlm.nih.gov/geo/)数据库当中的8个数据集(GSE19665、GSE84402、GSE60502、GSE45267、GSE64041、GSE39791、GSE76427以及GSE36376)和The Cancer Genome Atlas(TCGA,https://portal.gdc.cancer.gov/)数据库下载所有HCC基因的RNA测序数据,再应用R软件中的Limma软件包[4]对原始RNA测序数据标准化后进行差异分析,筛选出HCC组织与正常肝组织差异表达的基因。再使用R包“RobustRankAggreg”[5]对差异表达基因进行交叉整合,以|log2FC|≥1和P<0.05为有统计学意义。
1.2 对差异表达基因进行富集分析和蛋白质-蛋白质相互作用网络(PPI)分析
利用在线网站DAVID 6.8分析工具(https://david.ncifcrf.gov/)对上面筛选出的差异表达基因进行GO功能富集分析和KEGG通路富集分析,并进一步应用STRING数据库[7]进行PPI分析,将置信度得分设为0.9,构建得到的PPI网络再进一步导入Cytoscape[8],然后应用Cyto-Hubba插件中的Degree算法[9]进行分析,确定其中与发病机制密切相关的关键基因。
1.3 高风险组和低风险组HCC患者生存分析
通过TCGA数据库(包含有364例HCC患者的临床信息)获取HCC患者的总体生存时间(OS)(去除OS为0的患者),结合筛选出的HCC组织与正常肝组织差异表达基因,采用单因素Cox分析筛选出与HCC患者预后相关的基因。进一步将其中P<0.000 1与预后相关的基因再进行多因素Cox回归分析,根据基因表达值的线性组合构建预后风险模型,预测HCC患者的预后。计算公式为:预后风险评分=基因1的表达值×基因1的β1系数+基因2的表达值×基因β2系数+...基因n的表达值×基因n的βn系数[10]。根据预后风险评分的中位值将HCC患者分为低风险组和高风险组,应用Kaplan-Meier生存曲线比较两组患者的预后,使用R包“SurvivalROC”构建HCC患者ROC生存曲线。
2 结 果
2.1 HCC组织和正常组织差异表达的基因
在GEO数据库的8个数据集中下载HCC患者的RNA测序数据后,通过R软件进行标准化和差异分析,共筛选出452个HCC组织和正常组织差异表达的基因,包括242个下调基因和210个上调基因,同样的方法从TCGA数据库中共筛选出8 938个差异表达基因,包含1 546个下调基因和7 392个上调基因;对2个数据库中获得的差异表达基因使用R包交叉整合后,得到差异表达的基因400个,包括219个下调基因和181个上调基因。
2.2 差异表达基因的功能富集分析结果
对400个HCC组织和正常组织差异表达的基因进行GO功能富集分析,显示这些基因主要参与细胞分裂过程,排列在前10位的功能分别为细胞器裂变、核分裂、有丝分裂核分裂、对外源性刺激的反应、染色体分离、有机酸分解过程、羧酸分解过程、细胞对异生素刺激的反应、姐妹染色体分离、有丝分裂姐妹染色体分离;KEGG通路富集分析显示,400个差异表达的基因主要参与了与肿瘤代谢调节相关的途径,参与的前10个主要通路分别是细胞周期、DNA复制、药物代谢-细胞色素P450、视黄醇代谢、化学致癌作用、细胞色素P450对生物素的代谢、酪氨酸代谢、色氨酸代谢、脂肪酸降解、P53信号通路。
2.3 PPI分析筛选出差异表达的关键基因
对400个差异表达基因进行PPI分析,根据Degree算法进行排序显示,排名前10位差异表达基因分别为DNA拓扑异构酶Ⅱα(TOP2A)、细胞分裂周期蛋白20(CDC20)、细胞周期蛋白A2(CCNA2)、细胞周期蛋白B1(CCNB1)、核分裂周期蛋白80(NDC80)、细胞周期蛋白B2(CCNB2)、有丝分裂阻滞缺陷蛋白2(MAD2L1)、驱动蛋白超家族蛋白11(KIF11)、纺锤体检测蛋白(BUB1B)和细胞周期蛋白依赖激酶1(CDK1),即为HCC组织和正常组织差异表达的关键基因(图1)。
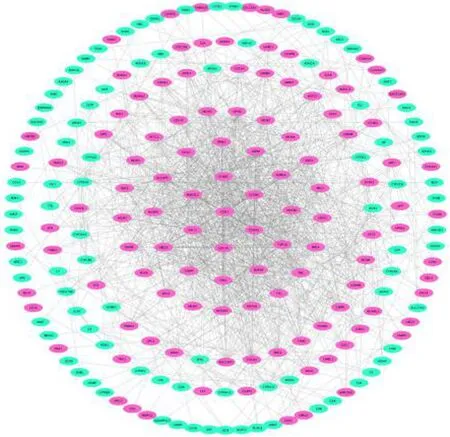
图1 HCC组织和正常组织差异表达基因的PPI分析图
2.4 HCC患者的生存分析
对TCGA数据库中获取的HCC患者OS以及400个差异表达的基因进行单因素Cox分析,总共筛选出与HCC患者预后相关的基因119个,其中P<0.000 1与预后相关基因40个,对40个基因再进行多因素回归分析,构建的预后风险模型中显示,醇脱氢酶4(ADH4)、组蛋白赖氨酸 N-甲基转移酶(EZH2)、剪接因子3B第4亚单位(SF3B4)、乙醛脱氢酶2(ALDH2)、血清对氧磷酶-1(PON1)、亚甲胺转移酶-环化脱氨酶(FTCD)、透明质酸介导的运动因子受体(HMMR)、核转运蛋白α2(KPNA2)、母系胚胎亮氨酸拉链蛋白激酶(MELK)、细胞因子信号传导抑制因子2(SOCS2)、分泌磷蛋白1(SPP1)以及甲状腺激素受体因子13(TRIP13)共12个基因与预后密切相关。所有患者的预后风险评分见图2A,预后风险评分的中位值为0.990,将风险评分>0.990的患者182例作为高风险组,风险评分≤0.990的患者182例作为低风险组,图2A中红色代表高风险组,绿色代表低风险组;HCC患者的生存状态分布图见图2B,显示高风险组患者的死亡数量显著多于低风险组,图中红色圆点代表死亡患者,绿色圆点代表存活的患者。Kaplan-Meier曲线显示,两组患者的OS存在显著差异(图3A),低风险组患者的预后显著好于高风险组(P<0.000 1);低风险组患者1、3、5年总体生存率分别为95.37%(95%CI=92.29%~98.56%)、8.75%(95%CI=71.06%~87.26%)及68.16%(95%CI=58.33%~79.66%);高风险组1、3及5年的总体生存率分别为70.41%(95%CI=63.81%~77.66%)、45.80%(95%CI=37.72%~55.61%)、28.78%(95%CI=20.00%~41.44%)。ROC生存曲线显示,HCC患者1、3、5年OS对应的AUC值分别为0.816、0.746和0.728(图3B)。

A:低风险组和高风险组患者的Kaplan-Meier曲线;B:HCC患者1、3和5年生存ROC曲线

A:HCC患者的预后风险评分分布图;B:HCC患者的生存状态分布图
3 讨 论
通过生物信息学的方法分析筛选肿瘤组织与正常组织差异表达的基因,并进一步进行PPI分析和生存分析,挖掘与肿瘤的诊断、治疗及预后密切相关的潜在生物学标志物,为后续的实验提供研究思路和方向,是目前新兴的一种行之有效的研究方法。
本研究首先对GEO数据库中的8个数据集和TCGA数据库中的RNA测序数据进行整合分析,筛选出HCC组织和正常组织差异表达的400个基因,包括219个下调基因和181个上调基因;GO功能富集显示,这些差异表达基因主要涉及有丝分裂、核分裂、染色体分离等功能。KEGG通路富集分析显示,这些差异表达基因主要参与调控细胞周期、DNA复制等通路。提示HCC的发生发展可能与调控细胞分裂和细胞周期的基因有关,或是与相关通路的激活等有关。通过对400个差异表达的基因进行PPI分析,发现了与HCC发生发展密切相关的关键基因10个,即CDK1、CDC20、CCNA2、CCNB2、CCNB1、MAD2L1、KIF11、BUB1B、TOP2A和NDC80,主要参与了细胞的有丝分裂。CDK1编码细胞周期蛋白依赖性激酶1,是CDK家族中细胞周期的关键调节激酶。相关研究表明CDK1的过表达与HCC的门脉侵袭、高甲胎蛋白水平以及预后不良具有直接关系[11]。
通过对TCGA数据库中HCC患者的OS进行单因素Cox分析和多因素Cox比例风险回归分析,发现与HCC预后相关的关键基因12个。研究发现,ADH4作为ADH家族的关键成员,其mRNA和蛋白的表达水平在HCC组织中均显著降低,与HCC患者的预后显著相关[12-13];ALDH2异常表达在许多疾病的发病过程当中发挥着关键作用[1];EZH2可通过上调间质标志物的表达和下调E-钙黏蛋白的表达来促进肿瘤转移[14],已经证明其过表达可与HCC的恶性进展和侵袭性表型密切相关[15];FTCD存在于哺乳动物细胞中,并且在肝脏的含量最高[16-17],已经被确定为原发性HCC的一种新型肿瘤标志物,在组织中的表达下调是HCC发生因素之一[18];HMMR最初被确定为透明质酸的受体,同时,HMMR是一种致癌蛋白[19];KPNA2是核转运蛋白家族的七个成员之一[20],在细胞的核质运输中起着至关重要的作用,其表达与HCC患者的不良预后相关[21];MELK是细胞周期依赖性蛋白激酶,在HCC组织中过表达可导致HCC早期复发和不良预后[22];PON1属于钙依赖性水解酶家族,血清中PON1的低表达总是与HCC中微血管侵犯相关[23],但目前尚未有研究发现PON1在HCC预后中的具体机制;SF3B4是剪接因子3b复合体的组成部分之一[24],其在HCC中高表达会导致HCC的肝内转移和不良预后[25-26];SOCS2属于细胞因子信号传导家族的蛋白质抑制因子,SOCS2低表达会导致肝细胞癌的侵袭性增加,并且SOCS2低表达往往造成HCC的不良预后[27];SPP1是在各种组织细胞中均表达的多功能细胞因子[28],目前认为是HCC早期复发和不良预后的潜在标志物[29];TRIP13蛋白可与甲状腺激素受体相互作用[30],主要参与细胞的分裂以及DNA的修复,研究发现TRIP13的高表达与包括HCC在内的各种癌症患者较差的预后有关[31]。
本研究针对与HCC预后密切相关的关键基因进行多因素Cox回归分析,构建预后风险模型,生存分析结果表明高风险组和低风险组患者的预后存在显著差异;低风险组患者1、3和5年总体生存率比高风险组高,提示低风险组患者的预后好于高风险组;构建的ROC曲线中HCC患者1、3和5年OS对应的AUC值分别为0.816、0.746和0.728,表明本研究所构建的模型预测效果较好。
本研究通过针对多个数据库中HCC患者的RNA测序数据进行生物信息学分析,从中筛选出与HCC发病机制和预后密切相关的关键基因,预测了这些关键基因可能参与的生物学途径和功能,并通过构建PPI网络和预后风险模型,对这些关键基因进行了预后分析,进一步验证了筛选出的关键基因与预后具有相关性。本研究中筛选出的与HCC发生和预后密切相关的基因,可能是HCC治疗的潜在靶点,但具体分子机制还需要进一步实验验证。
猜你喜欢
中老年保健(2022年1期)2022-08-17
江苏钢铁(2022年9期)2022-07-02
中学生数理化(高中版.高考理化)(2021年6期)2021-07-28
中国生殖健康(2020年4期)2021-01-18
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
中华老年多器官疾病杂志(2016年7期)2016-04-28
医学研究杂志(2015年5期)2015-06-10
医学研究杂志(2015年5期)2015-06-10
