
微囊型淋巴管畸形二例
2020-07-16 05:02宋翠豪许媛媛李承新
实用皮肤病学杂志 2020年3期
殷 广,巴 伟,宋翠豪,许媛媛,丁 潇,贺 赞,柏 佳,李承新,赵 华
临床资料
病例1,女,20 岁。因右大腿根部黄褐色及暗红色丘疹、水疱18 年,于2018 年7 月18 日就诊。患者2 岁时无明显诱因右大腿根部出现散在黄色小水疱,后逐渐增多,大小不一,水疱可自行消退或转变为丘疹,部分水疱可增大并呈暗红色,与衣物摩擦后可破溃。发病前后患处无明显自觉症状。曾就诊于当地医院,诊断为血管瘤,接受手术切除治疗,约1 个月后在手术部位再次出现黄褐色及暗红色丘疹、水疱,逐渐增多变大。既往体健,否认遗传病史及家族史。体格检查:各系统检查未见异常。皮肤科检查:右大腿根部可见一长约10 cm 手术瘢痕,周围可见多个大小不等的黄褐色及暗红色丘疹、水疱,直径0.2 cm ~0.8 cm,界限清楚(图1)。皮损组织病理示:表皮轻度增生,真皮乳头及真皮中部大量扩张的由单层内皮细胞形成的管腔,部分管腔内可见红细胞(图2)。免疫组化染色示:CD31、D2-40 阳性(图3)。诊断:微囊型淋巴管畸形。给予局部注射平阳霉素治疗1 次,后失访。
病例2,女,17 岁。因左髋部黄褐色及紫红色丘疹、水疱15 年,于2018 年6 月29 日就诊。患者15 年前无明显诱因左髋部出现紫红色水疱,曾就诊于某儿童医院,诊断为血管瘤,接受冷冻治疗,约1 个月后复发。9 年前,患者左髋部出现散在丘疹、水疱伴疼痛,在当地医院行超声多谱勒检查提示血管瘤,行手术切除,组织病理提示血管瘤。1 个月后,手术切口部位再次出现散在黄色小水疱,逐渐增多变大,部分水疱干瘪后形成丘疹或消退,少数水疱可融合并转为紫红色,伴疼痛,与衣物摩擦后可破溃。既往体健,否认遗传病史及家族史。体格检查:各系统检查未见异常。皮肤科检查:左髋部见长约10 cm 手术瘢痕,瘢痕旁见一直径约0.6 cm紫红色丘疹,界限清楚,周围散在黄褐色丘疹、水疱(图4)。组织病理示:表皮角化过度,不规则增生,真皮乳头层内扩张的由单层内皮细胞形成的管腔,部分管腔内可见红细胞,周围稀疏淋巴细胞浸润,真皮深部亦可见管腔样结构呈网状分布(图5)。免疫组化染色示:CD31、D2-40 阳性(图6)。诊断:微囊型淋巴管畸形。给予局部注射平阳霉素联合手术切除治疗,后失访。

图1 微囊型淋巴管畸形患者(例1)右大腿皮损
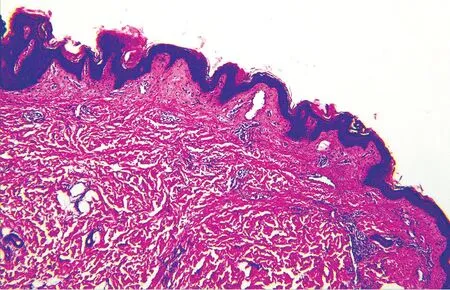
图2 微囊型淋巴管畸形患者(例1)皮损组织病理
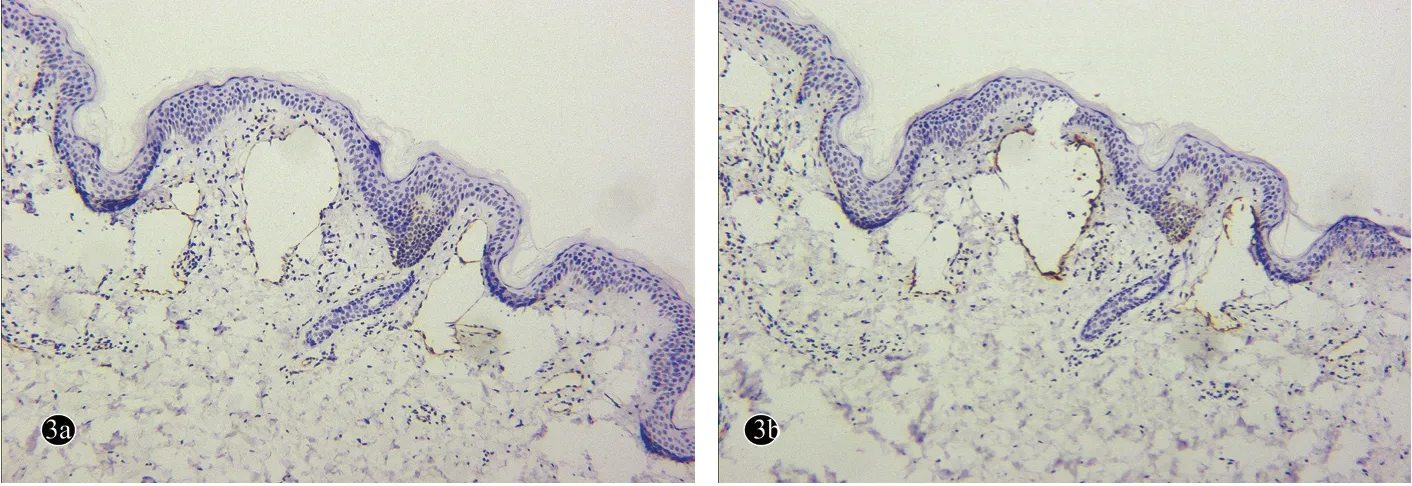
图3 微囊型淋巴管畸形患者(例1)皮损免疫组化(Envision 法 ×100)
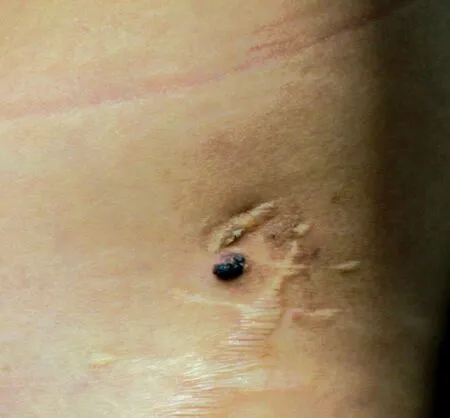
图4 微囊型淋巴管畸形患者(例2)左髋部皮损
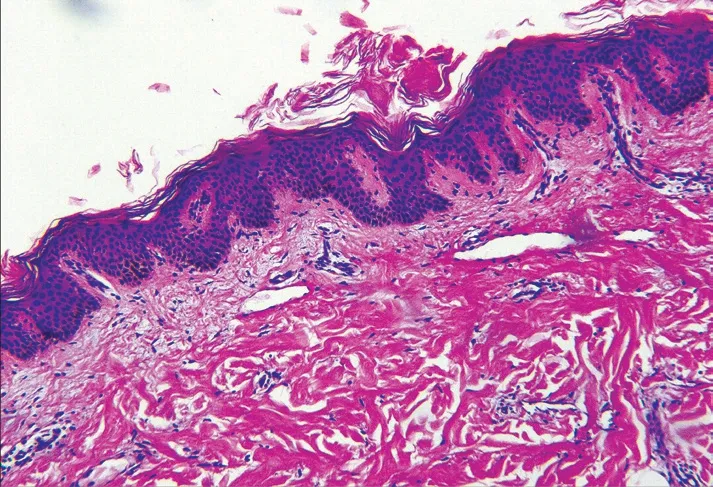
图5 微囊型淋巴管畸形患者(例2)皮损组织病理

图6 微囊型淋巴管畸形患者(例2)皮损免疫组化(Envision法 ×100)
讨论
淋巴管畸形(lymphatic malformation)是一种先天性的脉管畸形疾病,以往被称为淋巴管瘤,多见于幼儿,发病率为1/4 000 ~1/2 000[1,2]。其发病机制目前尚不完全清楚,一般认为是胚胎在淋巴管系统形成时期受到某种因素影响导致基因突变,从而引起淋巴管非恶性的异常生长和扩张,但功能表现正常。Luks 等[3]检测到淋巴管畸形伴有其他血管畸形或过度发育的综合征中存在体细胞PIK3CA基因突变。Blesinger 等[4]发现PIK3CA基因突变特异性地位于畸形的淋巴管内皮细胞,并能激活3-磷酸肌醇激酶(phosphoinositide 3-kinase,PI3K)/蛋白激酶B(protein kinase B,AKT)/哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)通路高表达。PI3K-AKT-mTOR 通路可能是淋巴管畸形非侵入性治疗的一个可行靶点。淋巴管畸形典型的组织病理学特点为真皮内单层内皮细胞组成的壁薄、形态不规则及大小各异的淋巴管腔内充满淋巴液,无内皮细胞数量的增加。根据囊腔的扩张大小,可分为3型:巨囊型、微囊型和混合型。巨囊型淋巴管畸形由1 个或多个体积≥2 cm3的囊腔构成,而微囊型淋巴管畸形则由多个体积<2 cm3的囊腔构成,二者兼有的则称为混合型淋巴管畸形[1]。本文报道2 例患者囊腔均<2 cm3,最终诊断为微囊型淋巴管畸形。该病一般出生时即有,但有时并不明显,出生后逐渐明显。65%~75%患者出生时即发现罹患此病,该病多在2 岁前发病[5],且皮损会随着患者生长发育成比例增长。它可发生在身体具有淋巴管网的任何部位,头部、颈部发病率最高,约占75%,腋窝约20%,腹股沟、纵隔、腹膜后次之,躯干及四肢最低[6]。本文报道2例均在躯干、四肢,不是本病的好发部位,较为罕见,这也是容易漏诊和误诊的原因之一。微囊型淋巴管畸形皮损可表现为皮肤黏膜上黄褐色、紫红色或暗红色的充满液体的小疱或者丘疹,不断出现的丘疹、水疱是其典型临床表现。临床治疗上常采用局部手术切除皮损,但此方法不能去除深在病灶,因此极易复发。当取局部皮损组织行免疫组化染色时可看到真皮内管腔内皮细胞胞膜CD31 和D2-40 均表达阳性。微囊型淋巴管畸形在临床上易被误诊为血管瘤,因此需与血管瘤进行鉴别。血管瘤一般在患者出生时或出生后均可发病,可分为增生期、消退期和消退完成期,大部分皮损可自行消退,临床表现一般为鲜红色或透出蓝色的单发丘疹,行局部手术切除后相同部位一般不再发生。血管瘤典型的组织病理学表现为真皮内多发扩张管腔,其内皮细胞异常增殖,囊腔大小不定,管腔内皮细胞免疫组化染色表现为CD31 阳性、D2-40阴性。
既往手术治疗是最主要的治疗手段,但随着硬化技术的发展,硬化治疗已经取代手术治疗成为淋巴管畸形的主要治疗方法,常用的硬化剂有平阳霉素、A 型链球菌制剂(OK-432)、强力霉素和聚桂醇等[7]。微囊型淋巴管畸形由于各囊腔存在分隔,单纯注射硬化剂治疗不能让药物到达每个囊腔,治疗效果欠佳。Churchill 等[8]的回顾性研究表明单纯使用硬化剂治疗微囊型淋巴管畸形有效率仅为46%。林文雄等[5]采用负压吸引联合硬化剂治疗8 例患儿,6 例治愈,2 例显效,疗效明显。王海珍等[9]采用多次注射博莱霉素治疗皮肤微囊型淋巴管畸形,平均治疗3.5次,30 例治愈,2 例显效,随访1 ~4 年未发现复发。近年来的研究表明西罗莫司作为一种mTOR 受体抑制剂对复杂淋巴管畸形,尤其是微囊型淋巴管畸形有很高的治疗价值。有作者报道眼眶的微囊型淋巴管畸形患者通过口服西罗莫司6 个月,病灶明显缩小[10]。也有研究发现外用西罗莫司治疗微囊型及混合型淋巴管畸形疗效确切,且不良反应仅为局部刺激,是一种安全有效的治疗方法[11],但还需大样本的随机对照临床试验验证。另外,还有一些研究通过联合用药[6]或与手术等方法相结合的形式[12]来提高治疗效果。更优化的标准治疗方案还需进一步探索。
猜你喜欢
中南药学(2022年8期)2022-11-19
南京医科大学学报(自然科学版)(2022年9期)2022-09-14
——淋巴管系统
科技视界(2022年7期)2022-04-13
组织工程与重建外科杂志(2022年1期)2022-03-05
医学前沿(2021年6期)2021-09-10
现代临床医学(2021年4期)2021-07-31
外科研究与新技术(2021年1期)2021-05-20
家庭科学·新健康(2020年6期)2020-07-06
江苏农业学报(2019年1期)2019-09-10
医学综述(2012年13期)2012-12-09
