
不伴MBTPS2与SAT1基因突变的棘状秃发性毛囊角化病1例
2020-07-16 05:02汤庄力王亚琦耿松梅牛新武肖生祥王晓鹏
实用皮肤病学杂志 2020年3期
汤庄力,王亚琦,肖 彤,王 添,耿松梅,牛新武,肖生祥,王晓鹏
棘状秃发性毛囊角化病(keratosis follicularis spinulosa decalvans,KFSD)是一种罕见的遗传性皮肤病,又称为Siemens 综合征。一般认为该病的遗传模式是X-连锁显性(XLD)与X-连锁隐性(XLR)遗传。该病由Siemens 等[1]在1925 年首次报道,典型临床表现为头发、睫毛及眉毛进行性脱发,部分患者可出现畏光、角膜营养不良等眼部症状[2]。截至目前,世界范围内关于该病的报道很少,PubMed 数据库中仅能搜索到50 篇。KFSD 被认为具有临床及遗传异质性。Gimelli 等[3]在2002 年提出SAT1基因参与KFSD的发病机制,而Aten 等[4]在2010 年通过基因连锁分析将该病的致病基因定位于MBTPS2基因。此外,Bellet 等[5]以及Castori 等[6]学者曾分别报道符合孟德尔常染色体遗传模式的KFSD 患者,但该遗传模式下的致病基因尚未被定位。本文的研究对象为KFSD 的散发患者,笔者通过检测患儿及其父母的MBTPS2基因及SAT1基因,意欲确定该患者的遗传模式,拓展该病的基因突变谱,为进一步阐明该病的发病机制,解释其临床及基因异质性奠定基础。
1 材料与方法
1.1 临床资料
患儿,男,8 岁。患儿出生后不久,双上肢逐渐出现毛囊性丘疹;1 年余前,开始出现弥漫性脱发,逐渐加重,残余毛发性状基本如常,未见卷曲、断发等改变。头皮、额部及双上肢可见弥漫分布的毛囊性丘疹,丘疹处未见毳毛。头皮弥漫性脱发,间有少量正常毛发生长,眉毛、睫毛以及其他部位皮肤基本正常(图1)。患儿自起病以来无畏光、视力异常等眼部不适;无皮肤瘙痒、疼痛等。患儿父母否认非近亲结婚,家族中无类似患者。
1.2 方法
1.2.1 皮肤活检术与外周血采集 向患儿及其父母充分告知病情特点以及相关检查的意义并签署知情同意后,局麻下于头皮枕部皮损处行皮肤活检术,所得标本用10%甲醛溶液固定,常规制片并苏木精-伊红(HE)染色。术后3 d 换药,7 d 拆线。拆线后手术切口处皮肤愈合良好,未见局部感染或血肿。
经肘正中静脉分别采集患儿及其父母外周血各2 ml,乙二胺四乙酸(EDTA)抗凝,-20℃深低温冻存。募集100 名健康志愿者(入组条件:男女不限,年龄18 ~40 岁,无系统性疾病或感染性疾病,凝血功能基本正常)作为健康对照,获取知情同意书后分别采集静脉血2 ml,编号后冻存备用。
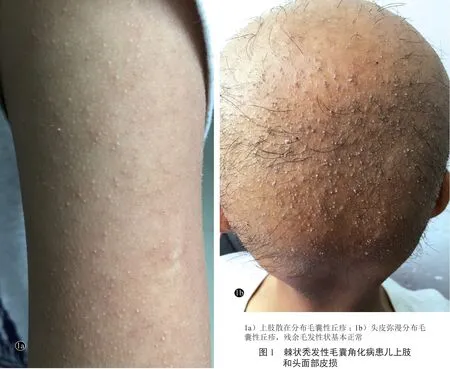
图1 棘状秃发性毛囊角化病患儿上肢和头面部皮损
1.2.2 外周血采集与基因组DNA 提取 室温缓慢熔化冻存的患者家系及健康对照外周血样本,使用全血基因组DNA 提取试剂盒[天根生化科技(北京)有限公司]提取基因组DNA 备用。
1.2.3 聚合酶链式反应(PCR)扩增MBTPS2与SAT1基因全部外显子及相邻内含子 采用Primer3.0 软件分别设计MBTPS2与SAT1基因全部外显子的上下游引物。每一个外显子的扩增体系为20 μl,体系内依次加入5 μl 焦碳酸二乙酯(DEPC)水,10 μl PCR 混合缓冲液,上下游引物各1 μl 以及DNA 模板3 μl。PCR反应条件为95℃预变性5 min;35 个扩增循环依次为变性(94℃,30 s),复性(56℃,30 s),延伸(72℃,1 min);72℃终末延伸5 min,4℃保存。
PCR 扩增产物在0.9%琼脂糖凝胶中电泳,全自动凝胶成像分析仪(JS-680B,上海培清科技有限公司)观察产物浓度以及特异性扩增情况。
1.2.4 DNA 序列分析 PCR 产物纯化后使用ABI3730XL 型全自动荧光DNA 测序仪(Applied Biosystems,CA,USA)测序,使用4peaks 软件观察测序图是否存在双峰,并将所测得序列与PubMed、HGMD Pro 以及dbSNP 数据库进行比对,分析是否有未报道的基因突变。
2 结果
2.1 皮损组织病理改变
HE 染色后光镜下观察,毛囊(枕部头皮)呈退行性变,毛干扭曲不完整,毛囊周围胶原增生,未见明显淋巴细胞浸润(图2)。结合临床考虑萎缩性毛周角化病。
2.2 基因测序
该例患者及其父母基因组DNA 在MBTPS2与SAT1基因均未见任何突变。

图2 棘状秃发性毛囊角化病患儿皮损组织病理
3 讨论
KFSD 与面部萎缩性毛发角化病(keratosis pilaris atrophicans faciei)、虫蚀状皮肤萎缩(atrophoderma vermiculatum)等疾病同属于萎缩性毛发角化病(keratosis pilaris atrophicans,KPA)[7],但皮损特点、累及部位与致病基因各不相同。KFSD 病因不明,主要表现为躯干及四肢的弥漫性毛发角化过度与头皮的瘢痕性脱发。该病患者虽然在出生后不久就可能出现眼部症状,但四肢与躯干的毛发角化与头皮的瘢痕性脱发常常始于青春期前后[9]。该病的组织病理学改变无特异性,可表现为明显的毛囊角化过度与毛囊口扩张,皮脂腺萎缩,毛囊和血管周围可见稀疏淋巴组织细胞浸润;毛囊周围纤维化可扩展至真皮网状层。
KFSD较难与毛囊性鱼鳞病- 脱发- 畏光 (ichthyosis follicularis,atrichia and photophobia syndrome,IFAP)综合征相鉴别。IFAP 综合征表现为毛囊性鱼鳞病、非瘢痕性非进展性脱发以及畏光,部分患者可以出现角膜炎、睑缘炎与牙釉质发育不良等临床表现,与KFSD 的部分临床表现相似。此外,MBTPS2基因突变亦可引起IFAP 综合征,因而有部分学者认为KFSD 是轻型的IFAP 综合征[2]。
KFSD 具有遗传异质性,MBTPS2与SAT1基因被认为是X-连锁KFSD 的致病基因。MBTPS2基因共包含11 个外显子,编码膜结合转录因子位点2 蛋白酶,对维持脂代谢稳态以及参与内质网应激反应具有重要意义。该蛋白酶失活会影响角质形成细胞的分化,进而造成皮肤组织病理改变[8,9]。SAT1基因编码亚精胺/精胺N 乙酰转移酶[10],Pietila 等[11]发现过表达该酶的小鼠模型会出现类似于人类KFSD 的临床表现,但具体机制尚不明确。而KFSD 在常染色体显性遗传模式下的致病基因尚未定位。
KFSD 准确的发病率尚不明确,未见有大规模流行病学报道。但国内外关于该病的报道均极少,截至目前,PubMed 数据库中关于该病的报道仅50 篇,涉及54 例散发或家系患者,累计发现MBTPS2基因突变23 个,其中错义突变21 个;国内仅报道了3 例KFSD 患者,并发现MBTPS2基因2 个新的突变[2,12,13]。
本例患者为散发病例,存在弥漫性毛发角化过度、头皮进行性瘢痕性脱发等体征,临床表现结合皮损组织病理改变符合KFSD 的诊断。但该例患者行基因学检测未发现MBTPS2基因与SAT1基因存在任何突变。
综上,笔者考虑该例患者可能存在其他未知的基因突变或基因部分片段缺失,具体的遗传模式有待于突变基因及位点的确定。家系全外显子扫描或全基因组扫描有助于发现潜在的基因学异常,对进一步阐明KFSD 的发病机制以及基因异质性有重要意义,相关研究仍在进行中。
猜你喜欢
数学学习与研究(2021年18期)2021-08-06
皮肤病与性病(2021年3期)2021-07-30
感染、炎症、修复(2021年1期)2021-07-28
西南农业学报(2021年1期)2021-05-25
爱你(2019年41期)2019-11-16
文萃报·周二版(2019年37期)2019-09-10
小学生导刊(2018年13期)2018-06-29
中学生理科应试(2017年6期)2017-09-27
东方教育(2017年14期)2017-09-25
课程教育研究·学法教法研究(2016年1期)2016-03-17
