
萤石的晶体化学基因特性量化计算与分子动力学模拟
2020-07-14 06:34史新章张金山王介良曹永丹董红娟
金属矿山 2020年6期
史新章 曹 钊,2,3,4 张金山 王介良 曹永丹 董红娟
(1.内蒙古科技大学矿业研究院,内蒙古包头014010;2.广东省资源综合利用研究所,广东广州510650;3.稀有金属分离与综合利用国家重点实验室,广东广州510650;4.内蒙古自治区矿业工程重点实验室,内蒙古包头014010)
氟(F)是新能源、新材料、国防、光电、化工等领域 必不可少的化学材料[1]。萤石(CaF2)作为氟的主要来源,其含氟量高达48.67%,是地壳中最重要的含氟矿物相,已被列为我国的战略性矿产资源[2-3]。
浮选是矿物分离的主要方法之一,浮选药剂在矿物颗粒暴露面上吸附的稳定性差异决定了矿物分离的效果。萤石常与稀土矿、钨矿、方解石等矿物伴生,脂肪酸类捕收剂是其浮选分离的常用药剂[4]。由于萤石与稀土矿、方解石、白钨矿等矿物的可浮性相近,其浮选分离是行业内难题[5]。因此,研究萤石的晶体化学、晶面性质以及药剂在萤石不同晶面上的吸附构型和吸附能等矿物基因特性对研发新型浮选药剂、实现萤石高效浮选分离有重要的指导意义。
分子模拟方法可以从原子角度去研究矿物晶体和晶面性质,预测药剂分子在矿物表面的吸附构型和作用方式,定量计算药剂在矿物表面作用过程中的能量及电荷变化等基因特性[6-8]。与传统的浮选试验相比,分子模拟技术具有耗时短、成本低、易操作等优点,已经成为矿物加工与分选领域工作者研究吸附机理、设计开发新型浮选药剂分子的重要手段之一[9]。Gao等[5,10]基于密度函数理论(DFT)计算了几种萤石晶面的表面能和表面断裂键密度等矿物基因特性,发现表面能与萤石的断裂键密度呈线性相关。Zhu等[6]凭借量化计算进一步探究了在石英(101)面上新型石英捕收剂α-BLA(α溴代月桂酸)的作用机制,观察到Ca(OH)+的活化影响是因为其在石英表面与α-BLA间发挥了“桥联”作用。Maldonado[11]等利用第一性原理计算模拟预测了萤石的表面稳定性,发现萤石(111)面表面能最低,是萤石最稳定的晶面。前人研究成果表明,模拟结果与实际微浮选试验结果基本一致,证明了分子模拟技术研究矿物晶体化学基因特性的有效性与实用性。
萤石的浮选分离通常在水溶液中进行,水在NaOL水解和萤石颗粒的溶解中起重要作用。而大多数模拟研究在真空系统中进行,水溶液对液固界面的影响被简化甚至忽略,导致模拟的结果不具说服力。本文利用DFT计算了萤石晶体的(111)面、(110)面、(311)面和(100)面4个常见解理面和暴露面的断裂键密度,对萤石的晶体化学、晶面性质等基因特性进行表征;通过在水溶液系统中的分子动力学(MD)模拟,研究并阐述了NaOL在不同萤石晶面上的吸附行为以及相关的吸附构型和相互作用能,结合相关的量化计算以期探明NaOL在萤石晶体不同暴露面上的吸附机理。
1 萤石结构与研究晶面确定
1.1 萤石晶胞结构
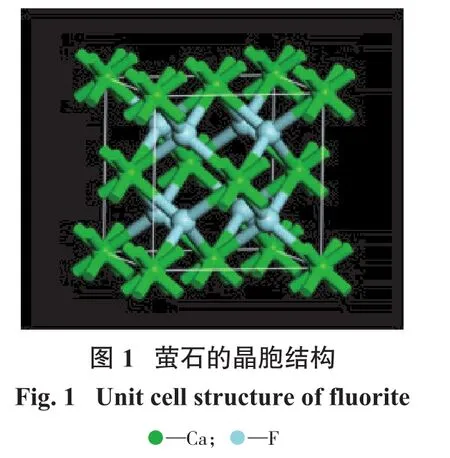
萤石的晶胞结构和参数由美国矿物学家晶体结构数据库获得[12],其晶胞参数:a=b=c=0.546 31 nm,α=β=γ=90°,空间群为Fm3 m,Z=4,属于立方晶系。萤石的晶胞结构如图1所示。Ca2+在萤石晶体结构中以立方紧密方式堆积,四面体的空隙位置被F-占据。Ca2+与附近的8个F-结合成8配位,形成Ca-F8立方体,F-与附近的4个Ca2+结合成4配位,形成正四面体[13]。Ca—F键的键长为0.236 6 nm。Ca—F键的离子键分数为89.14%,化学键的离子键分数大于50%,可认为该化学键为离子键,因此Ca—F键为强离子键[5]。
1.2 研究晶面的确定
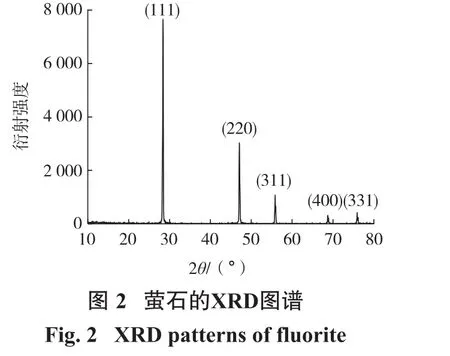
根据萤石晶体的X射线衍射(XRD)谱图(图2)确定主要强峰,找出这些主要强峰对应的晶面。衍射峰的前 4个强度对应的(111)、(220)、(311)和(400)面更容易通过机械粉碎和研磨获得。
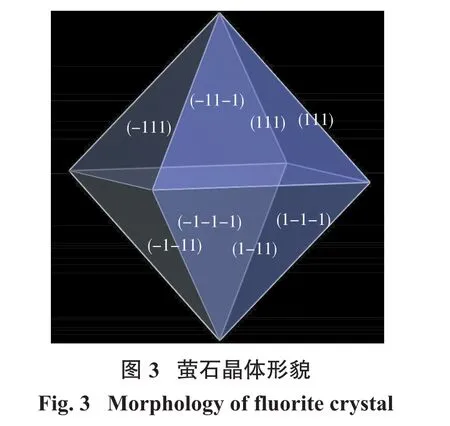
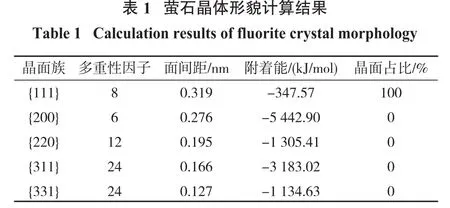
附着能(AE)模型是基于Hartman和Perdok提出的PBC理论[14-16]。运用PBC理论可以确定晶体薄片的附着能Eatt,晶面的附着能越低,其生长速率越小,因而越容易成为晶体形貌中最常见的晶面[17]。本文基于Material Studio 2017软件的Morphology模块计算了萤石晶体的形貌,计算结果如图3和表1所示。
由图3和表1可知,{111}晶面族的晶面附着能最小,晶面占比为100%,结合XRD测试结果,最终确定(111)、(110)、(311)和(100)面为可研究晶面((110)面,即(220)面的平行晶面;(100)面,即(400)与(200)面的平行晶面)。
2 模拟方法
2.1 计算模块的选择
利用Materials Studio 2017软件中的CASTEP和Forcite模块进行相关计算。CASTEP模块用于密度泛函计算,包括萤石晶体结构与表面模型的优化和能量计算等任务。Forcite模块用于油酸钠与萤石不同晶面的分子动力学模拟,该模拟在模块中的COMPASSⅡ(原子模拟研究的凝聚相优化分子势)力场下进行。COMPASSⅡ力场包括常见的有机分子和无机材料,包括金属、金属卤化物、铝硅酸盐和金属氧化物等[18],这使其可以合理地应用于构建的模型。
2.2 晶胞优化
通过MS2017中的Build Crysatls模块对萤石晶胞建模,然后采用CASTEP模块对其进行几何优化,其中交换相关泛函选择广义梯度近似Wu-Cohen(WC)方法[19]。几何优化的收敛参数设置为:最大原子间作用力为0.3 eV/nm,最大原子位移为1×10-4nm,各个原子的能量收敛误差小于1×10-5eV,最大原子内应力0.05 GPa。SCF收敛精度设置为Fine,其收敛误差不超过1×10-6eV,赝势采用OTFG Ultrasoft,平面波截断能Energy cutoff设置为394.6 eV,k点取样密度为6×6×6,k点取样间隔separation在几何优化过程中设置为4×10-3/nm。上述参数在后续计算中若无特殊情况不做更改。
优化后的晶胞参数为:a=b=c=0.551 98 nm,α=β=γ=90°,空间群为Fm3m。将优化后的晶胞参数与实验值对比发现误差小于3%,表明参数设置合理,该模型可以用于后续计算。
2.3 矿物表面模型与DFT计算
将优化后的晶胞通过supercell建立2×1的超晶胞,沿着不同的方向对其切割,形成(111)面、(110)面、(311)面以及(100)面,在不同晶面上方建立真空层得到各自的表面结构。随后在CASTEP模块中选择WC泛函计算萤石不同晶面的表面能。表面层和真空层厚度会对表面结构的稳定性造成影响,其稳定性可由表面能表征,越低的表面能代表着表面结构稳定性越好[6]。当表面层和真空层厚度继续增加时,表面能会趋于稳定,所以对其进行收敛性测试为后续计算构建最稳定的表面模型[20]。
分别采用式(1)和式(2)计算不同晶面的断裂键密度和表面能[21]。
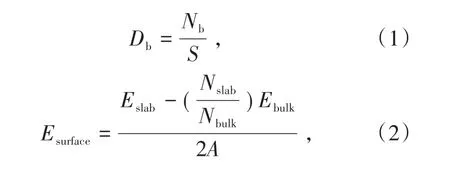
其中,Db和Nb分别为晶面断裂键密度和某晶面单胞区域内的未饱和键(断裂键)个数;S为该晶面上单胞区域的面积;Eslab和Ebulk分别为表面结构和单胞的总能量;Nslab和Nbulk分别为表面结构和单胞的总原子数;A为表面结构沿Z轴方向的面积;2表示表面结构沿Z轴方向有上下2个表面。
2.4 NaOL溶液模型
借助MS的Amorphous Cell模块构建NaOL溶液模型,如图4所示。力场设置为COMPASSⅡ,采用力场分配电荷平衡法(force-field-assigned)计算原子电荷。NaOL溶液模型的尺寸设置为3.9 nm×3.9 nm×3.6 nm、3.8 nm×3.9 nm×3.6 nm、4.1 nm×3.9 nm×3.4 nm、3.9 nm×3.9 nm×3.5 nm。

2.5 分子动力学模拟细节
将优化好的萤石单胞切割,建立(111)面、(110)面、(311)面和(100)面,使用supercell将UV值设置为10×10、7×10、6×10和10×10的表面单元,算法采用smart,收敛水平为10-4kcal/mol,然后对每个晶面进行几何优化以确定底部原子。在求和方法里将van der Waals设置为Atom-based,electrostatic设置为Ewald,Atom-based求和的截止距离为1.55 nm,采用力场分配电荷平衡法计算原子电荷。为结合NaOL溶液箱,在优化表面的基础上,建立真空层厚度为0的萤石表面模型。按照与之前相同的参数设置,再次对由矿物表面和NaOL溶液箱组成的复合模型进行几何优化。为获得药剂与矿物表面吸附平衡状态下的最佳位置,进行MD模拟以使能量收敛。在MD模拟之前,(111)面、(110)面、(311)面和(100)面的矿物-试剂复合物的最终模型尺寸分别设置为3.9 nm×3.9 nm×14.7 nm(总原子数:7 380)、3.8 nm×3.9 nm×14.4 nm(总原子数:7 680)、4.1 nm×3.9 nm×13.9 nm(总原子数:7 740)和3.9 nm×3.9 nm×14.2 nm(总原子数:7 980)。
MD模拟使用NVT系综(恒定体积和恒定温度),thermostat设置为Velocity Scale,时间步长为1 fs,模拟时间为200 ps,使得整个系统的能量达到平衡。接着将thermostat设置为Nose,Q比率为0.01,温度为298 K,时间步长为1 fs,总模拟时间为1 000 ps。矿物表面与药剂相互作用的相对亲和力由相互作用能表示,由下式计算[18]:
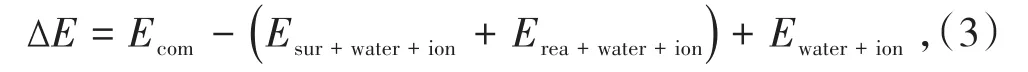
式中,Ecom为优化后复合模型的总能量;Erea+water+ion和Esur+water+ion分别表示没有矿物表面的系统和没有试剂的系统能量;Ewater+ion是钠离子和水分子的能量。相互作用能的值(ΔE)表示吸附系统的稳定性。相互作用能值越负表明矿物表面和药剂之间的相互作用越强,吸附过程可以自发地进行,而作用能为正则意味着药剂与矿物表面的相互作用较弱,吸附过程不能自发地进行。基于MD模拟的结果,使用Forcite模块进一步计算了沿晶体表面Z轴NaOL中代表性原子(C和O原子)的浓度分布,以阐明吸附构型。
3 结果与讨论
3.1 萤石表面晶体特性分析
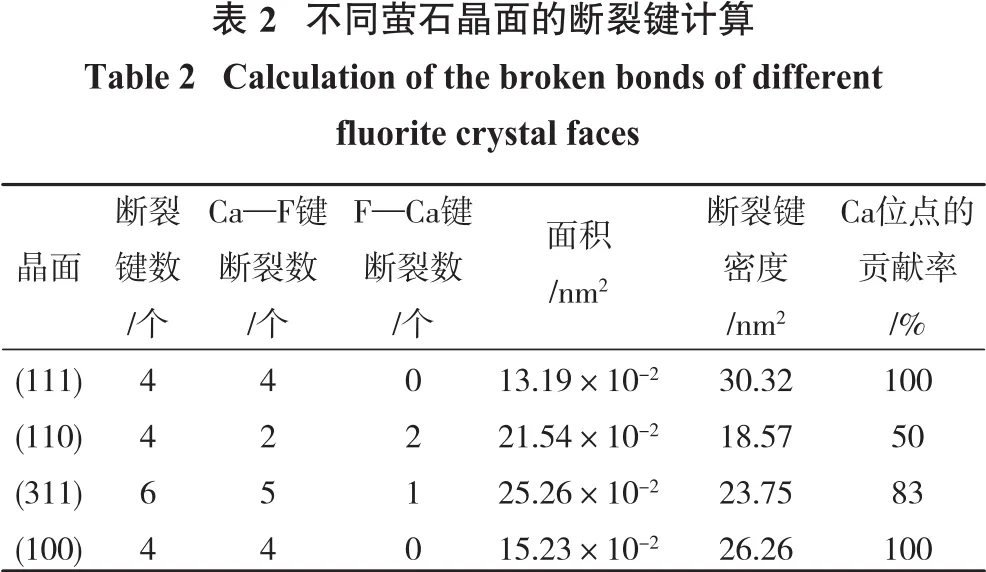
矿物的晶面性质受其晶体化学的影响,特别是对于细小的矿物颗粒。随着矿物颗粒粒径的减小会暴露更多的晶面,图5为萤石不同晶面的结构及其断裂键情况。由晶体的周期性可知在(111)面上存在不饱和的Ca原子,其具有4个Ca—F断裂键。除了Ca—F断裂键以外,在(110)面上还观察到有2个不饱和的F原子,其具有2个F—Ca断裂键。在(311)面上观察到1个不饱和的F原子,其具有1个F—Ca断裂键。(100)面的断裂键情况与(111)面相似,也具有4个Ca—F断裂键。NaOL主要是与萤石晶面的Ca位点吸附结合,矿物晶面上的Ca位点越多其活性越强,与药剂的反应程度越高[5],由此可得知(111)和(100)面活性较强。

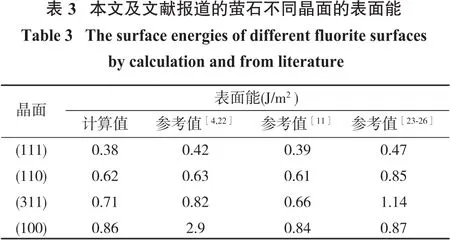
表2和表3为萤石不同晶面的表面能和断裂键密度的计算结果。由表2和表3可知,萤石不同晶面的断裂键密度(Db)按以下顺序递减:(111)面>(100)>(311)面>(110)面。通过分析表面能、断裂键密度和面间距等矿物基因特性可知,虽然(100)和(111)面都具有100%的Ca—F断裂键,但是(100)面的面间距小于(111)面,其表面能大于(111)面,因此(111)面为萤石的最稳定解理面;(100)面由于其表面能较高不易暴露,(110)面比(311)面更容易暴露,上述结论与前人的研究结果一致[5,11]。
3.2 相互作用能比较
图6为NaOL在不同萤石晶面上吸附前后的复合模型。NaOL捕收剂被溶解成油酸阴离子(去质子化的RCOO—基团)和钠离子(Na+)。由图6可知,在经过MD模拟后,几乎所有的油酸阴离子都吸附在(111)、(311)和(100)面上,这些晶面的水膜被破坏,其中(111)面的吸附效果最为显著,而萤石(110)面几乎没有油酸阴离子吸附。

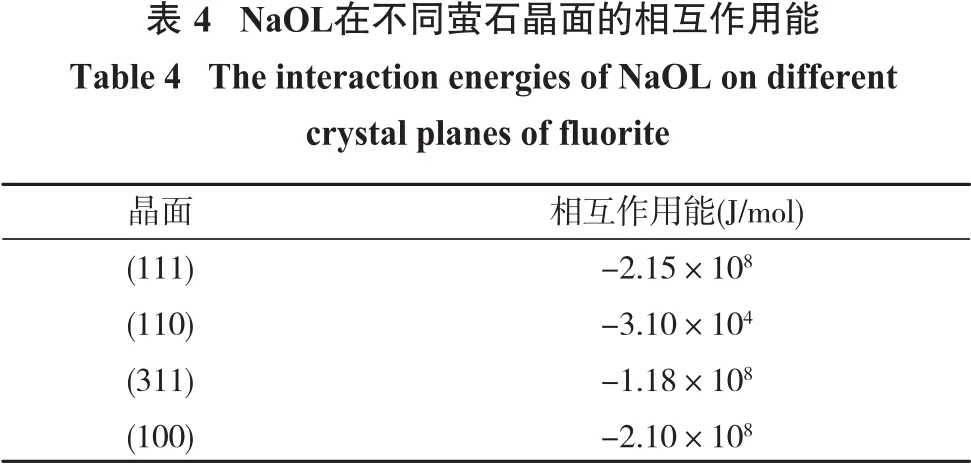
药剂在矿物表面上的相互作用能越负,药剂与矿物表面的亲和力越高,表4为NaOL与不同萤石晶面的相互作用能。
由表4可知,NaOL与萤石(100)面的相互作用能和(111)面接近,但比(311)面的要负得多,NaOL几乎不与(110)面发生吸附作用。结合前文中的有关结论,NaOL更倾向于与最稳定的(111)面吸附结合。
3.3 吸附构型性质
NaOL在不同萤石晶面上的吸附构型如图7所示。
由图7可知:几乎所有的NaOL以双核双配位构型与(111)面上的Ca位点结合,这是油酸盐与含钙矿物相互作用的最稳定构型[5];萤石(110)面有50%的F—Ca断裂键,大量水分子通过与F原子形成氢键吸附在(110)面导致其被较厚的水膜覆盖,高亲水性使得NaOL在(110)面的吸附变得困难;在NaOL与(311)面的配合物结构中发现了单核单配位、双核单配位和双核双配位3种吸附构型,由于其表面有17%的F—Ca断裂键通过与水分作用形成了氢键,水分子比(111)面更接近(311)面;NaOL与(100)面的配合物结构中发现单核单配位和双核双配位2种吸附构型,其中双核双配位构型占主导地位。
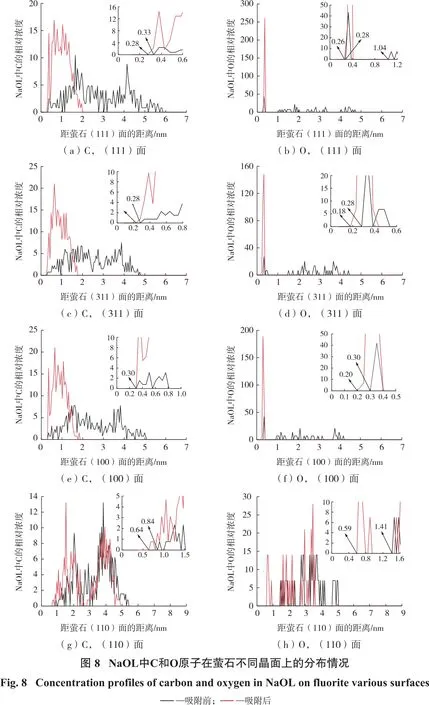
为了进一步阐明吸附构型,计算了NaOL中代表性原子(C和O原子)沿晶面Z轴的浓度分布,以定量说明NaOL在萤石不同晶面上的分布情况,如图8所示。
由图8可知:NaOL在吸附后,C和O原子与萤石晶面间的距离变化明显,并在萤石(111)、(311)和(100)面上有规律地分布;其中C原子分布在距萤石(111)面约0.27~2 nm的范围内,而O原子分布在距(111)面约0.20~0.4 nm的范围内,最强的原子键长为0.20 nm,在(311)和(100)面也发现了同样的规律。上述结果表明NaOL在萤石(111)、(311)和(100)面发生稳定吸附;萤石(110)面附近没有C和O原子分布,而且C和O原子的浓度及其与萤石(110)面间的距离在吸附前后没有发生较明显的变化,结合相互作用能与吸附构型的分析,表明NaOL几乎不与(110)面发生作用。

4 结论
(1)通过X射线衍射分析与MS晶体形貌计算,确定了萤石的4个常见暴露晶面,分别为(111)、(110)、(311)和(100)面;通过对萤石不同晶面的表面能比较,发现(111)面的表面能最小,表明(111)面是萤石最稳定的解理面。
(2)通过DFT计算表面断裂键分析,萤石晶面的各向异性与其原子种类与排布有关。(111)和(100)面具有100%的Ca—F断裂键密度,对NaOL有很强的亲和力,吸附作用较强;(110)面和(311)面随着萤石被粉碎并研磨成更细小的颗粒时会暴露出来,由于其分别含有50%和17%的F—Ca断裂键而亲水,从而降低对NaOL的亲和力,吸附作用较弱。
(3)通过相互作用能以及原子浓度分布分析,OL-阴离子与(111)面的相互作用能绝对值最大,以双核双配位构型优先与其相互作用并产生稳定吸附;OL-阴离子与(110)面的相互作用能绝对值最小,NaOL几乎不与其发生作用。
猜你喜欢
军民两用技术与产品(2022年1期)2022-06-01
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
矿产勘查(2020年7期)2020-12-25
物理实验(2019年7期)2019-08-06
航空材料学报(2019年2期)2019-04-15
西安工业大学学报(2018年6期)2018-02-13
北京航空航天大学学报(2017年10期)2017-04-20
文物保护与考古科学(2016年4期)2016-05-17
铜业工程(2015年4期)2015-12-29
云南民族大学学报(自然科学版)(2015年4期)2015-11-14
