
聚噻吩液晶/多壁碳纳米管复合材料的制备及其性能研究
2020-07-11 02:40屈琦超刘克建何意林
化学反应工程与工艺 2020年5期
屈琦超,刘克建,何意林
1.重庆工业职业技术学院化学与制药工程学院,重庆 401120;
2.兰州交通大学化学与生物工程学院,甘肃 兰州 730070
近年来,液晶材料已在图像显示、温度探测、气体检测及医学诊断等领域[1-3]得到广泛的应用。虽然针对高分子液晶材料的研究时间不长,但其应用性强,具有高强度、高模量、成型收缩率低、膨胀系数低、介电性好和耐腐蚀性强等优良性能[4]。然而,液晶高分子还存在着液晶相区间短、稳定性差、接缝强度低和经济性差等不足,限制了其应用领域。因此,开发合适的掺杂材料进行复配,优化高分子液晶材料性能,具有重要的现实和经济意义。
同时大多数液晶聚合物乃至液晶单体由于其结构关系存在熔点高、刚度小、极性大和加工成型困难等不足[5]。为了克服单一材料性能的不足,学者们针对其特点进行了各种方法的改进,如在其分子链上通过嵌段或接枝手段同时引入液晶段和非液晶段,使两相材料相互粘接,增强互溶性,改善了材料的性能[6]。通过熔融热致液晶共聚酯与双酚A、聚碳酸二苯酯制得液晶嵌段共聚物,即为液晶聚合物/聚碳酸酯共混体系[7],其性能较单一材料明显改善。为提高液晶材料的可塑性及化学性质稳定性,聚对苯二甲酰对苯二胺/聚酰胺、聚苯并噻唑/聚苯并咪唑和芳香族聚酰胺/聚氯乙烯等高分子复合材料等先后被开发出来[8],但至今未能工业化应用。此外,液晶高分子可在未聚合前加入第二相材料(非液晶材料),最后聚合形成“原位复合材料(in-situ Composites)”,它是当前应用最广泛的一类新型液晶复合材料[9-10],如液晶聚合物/聚醚砜[11]、液晶聚合物/聚砜[12]、液晶聚合物/聚醚酮、液晶聚合物/聚酰胺[13]、液晶聚合物/聚碳酸酯[14]和碳纳米管/缩聚液晶[15]等液晶复合材料,均具有各自材料的优良性,并得到了互补和关联,改善了综合性能。
因此,以材料的开发优化为目的,本工作在合成一种新型噻吩类小分子液晶单体的基础上,采用原位聚合法使其与经过结构修饰的多壁碳纳米管发生聚合反应制备了一种新型的液晶高分子复合材料,并优化了合成工艺。采用红外光谱(FT-IR)、核磁共振氢谱对该材料的结构进行了表征,通过透射电镜和热致偏光显微镜分析等方法对其液晶性能进行了分析,发现该种方法能使分散相与基体相结合,在熔融状态下依然均匀分布,整齐地排列在液晶相的涡流中,增大了液晶相的范围。
1 实验部分
1.1 材料的制备
1.1.1 多壁碳纳米管的结构修饰
将1.2 g多壁碳纳米管加入到装有由98%浓H2SO4与68%浓HNO3(其中浓硫酸45 mL,浓硝酸15 mL)混合溶液的三口烧瓶中,加热到90 ℃,搅拌反应2 h,反应结束,冷却至室温,离心得到黑色粉末状固体。先用丙酮洗涤2~3次,再用蒸馏水洗涤至pH值为中性,干燥,得到改性后的多壁碳纳米管,备用。
1.1.2 4-(2-噻吩甲亚胺基)苯酚的合成
参考文献[16]制备4-(2-噻吩甲亚胺基)苯酚,其合成路线如图1所示,在250 mL三口烧瓶中加入8.00 g 4-氨基苯酚(0.08 mol)及100 mL无水乙醇,磁力搅拌,待4-氨基苯酚完全溶解以后,氮气保护下滴加8.96 g 2-噻吩甲醛(自制,0.08 mol),避光条件下回流反应4 h后,趁热抽滤。滤液冷却后有大量浅黄色晶体析出,抽滤得到粗产品,无水乙醇重结晶即得浅黄色晶体。
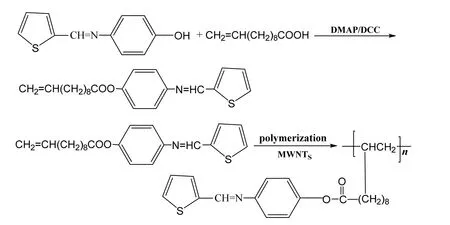
图1 聚4-(噻吩-2-亚甲基氨基)苯酚的合成路线Fig.1 Synthesis route of poly (4-(thiophene-2-methylamino) phenol
1.1.3 4-(噻吩-2-亚甲基氨基)-10-十一烯酸苯酯的合成
参考文献[17]制备,向500 mL单口烧瓶中先加入150 mL溶有4.06 g 4-(2-噻吩甲亚胺基)苯酚(0.02 mol)的二氯甲烷溶液,再在不断搅拌下加入0.10 g(0.8 mmol)4-二甲氨基吡啶(DMAP)和2.78 g(0.02 mol)十一烯酸,超声处理至所有固体完全溶解后,加入100 mL溶有4.16 g(0.02 mol)N,N-二环己基碳酰亚胺(DCC)的二氯甲烷溶液,外接干燥管,室温超声搅拌反应15 h后,减压抽滤,滤液减压蒸发除去溶剂,无水乙醇重结晶即得黄色晶体状目标产物。
1.1.4 MWNTs/液晶高分子复合材料的制备
将0.35 g多壁碳纳米管(MWNTs)、6.02 g 4-(噻吩-2-亚甲基氨基)-10-十一烯酸苯酯和2 g聚乙烯吡咯烷酮加入到研钵中研钵研磨混合均匀,再将混合物加入到装有150 mL无水乙醇/1,2二氯乙烷(两者体积比2:1)溶液的圆底烧瓶中,超声处理1 h后,加入以过氧化苯甲酰1.50 g,微波条件下于60 ℃剧烈搅拌反应一定时间即得聚4-(噻吩-2-亚甲基氨基)-10-十一烯酸苯酯与多壁碳纳米管复合材料,其动力黏度为90 Pa·s。
2 结果与讨论
2.1 反应温度和加热方式对目标反应的影响
反应时间为25 min,分别采用电热套加热和微波反应器加热方式,考察不同反应温度对目标反应的影响,结果见表1。由表可知,采用电热套加热方式时,温度低于100 ℃不能引发反应,在100~110 ℃能引发反应,但反应体系不稳定,易碳化,副反应较多。由此可知,低温不能引发反应,高温副反应较多,温度的调节不能改善反应结果。改用微波反应器加热反应,温度在50 ℃时引发反应,反应体系变清亮、黏稠速度慢。温度升高到60 ℃,黏稠速度明显提高,反应25 min体系动力黏度为90 Pa·s,随着反应温度的升高,在70和80 ℃,反应体系颜色加深,副反应较多。
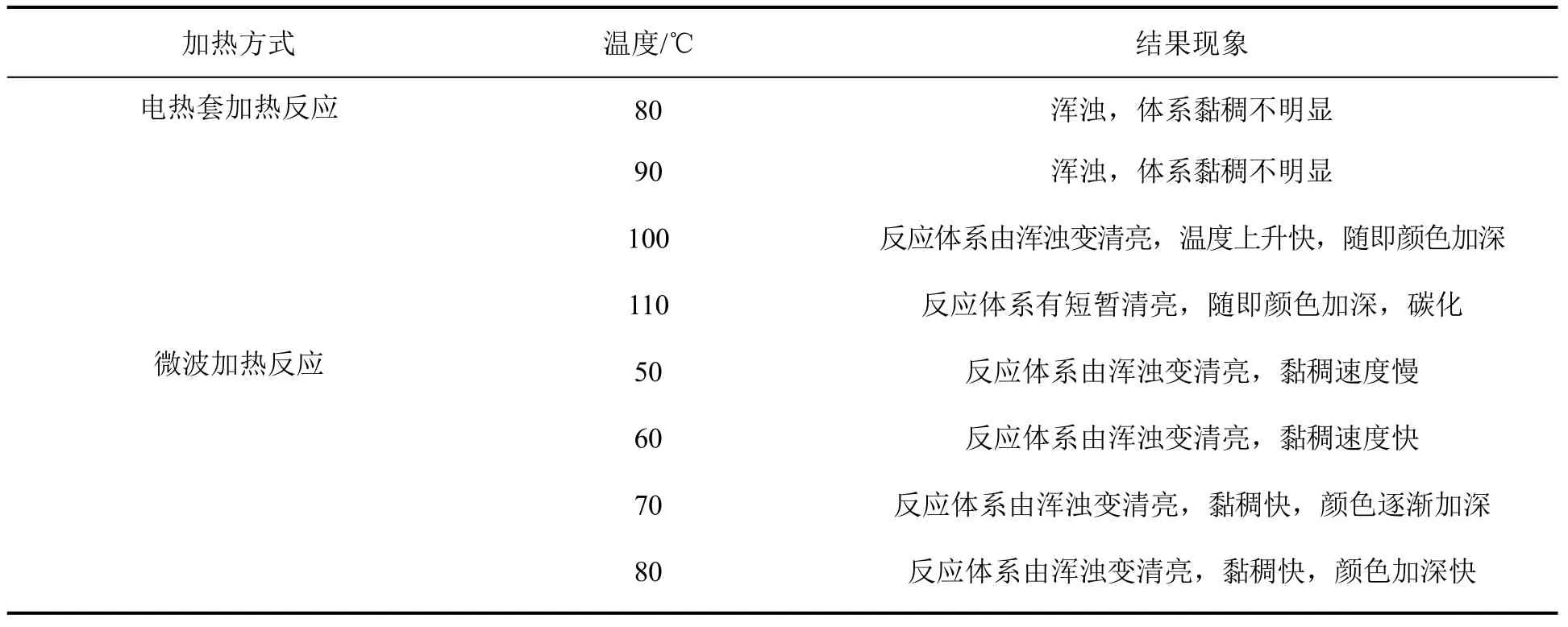
表1 加热方式及反应温度对目标反应的影响Table 1 Influence of heating mode and reaction temperature on target reaction
综上所述,普通电热套加热反应需高温引发,但伴随副反应较多,微波反应器反应,低温可进行,且副反应少。
2.2 单体与改性多壁碳纳米管比例对目标反应的影响
在微波反应器中,考察单体4-(噻吩-2-亚甲基氨基)-10-十一烯酸苯酯与改性多壁碳纳米管的不同质量比对目标反应的影响,结果见表2。

表2 单体比例对目标反应的影响Table 2 Influence of monomer ratio on target reaction
由表2可知,产品随着单体与多壁碳纳米管质量比的增大,产品颜色逐渐加深,熔融状态下,碳纳米管颗粒从分离状态到融为一体,液晶相漩涡从漩涡明显到有序排列(见图8),再到漩涡不明显。单体比例太大,分散系不足,不能充分体现复合材料性能;比例太小,分散系剩余,团聚沉积在一起,影响液晶相,漩涡不明显,有序性差。因此,质量比为17:1较佳。
2.3 引发剂与单体比例对目标反应的影响
在微波反应器中,热引发温度为60 ℃,选用有机过氧化物中温引发剂(30~100 ℃)。由于单体分子量较大,双键π电子活泼性降低,聚合反应较慢,在引发剂过氧化苯酰的刺激下,于微波条件下(避免了单体的分解),充分将能量转化为分子内能,引发反应。考察引发剂和单体质量比对目标反应的影响,结果见表3。引发剂含量低时,不能引发反应或者反应速率较慢,随着引发剂含量的增加,反应速率加快,甚至副反应会增多,由表可知,当引发剂与单体的质量比为2.5:10时,反应较好。

表3 引发剂比例对目标反应的影响Table 3 Influence of initiator ratio on target reaction
2.4 表征分析
2.4.1 改性前后MWNTs的红外光谱分析
一般来说多壁碳纳米管直接进行掺杂制备复合材料,往往分散性、稳定性都较差,通过对其进行改性,引入基团,两者之间的结合犹如氢键结合,从而更加牢固。盐酸以及混酸处理后多壁碳纳米管的红外光谱如图2所示。由图2可知,由单一盐酸进行改性的多壁碳纳米管,在波数为3 421,1 561,1 232以及3 000~2 800 cm-1存在吸收峰,其主要是由—OH,C=O,C—C—O和—CH—产生的特征峰。但在硫酸与硝酸混合物中进行改性后,在3 446,1 633和3 660~3 000 cm-1出现了特征吸收峰,分别代表—OH,C=O,和—COOH的振动峰。由图2还可以看出,经浓硫酸/浓硝酸混酸处理多壁碳纳米管特征峰面积更大,说明碳纳米管氧化程度更强,而这有利于复配时的结合。
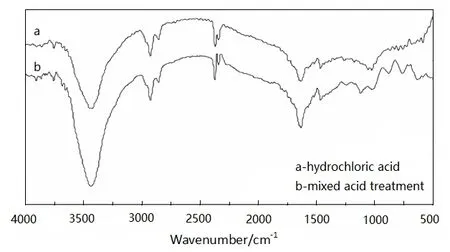
图2 多壁碳纳米管在盐酸及混酸处理后的红外对比Fig.2 Infrared comparison of MWNTs of hydrochloric acid and mixed aced treatment
2.4.2 多壁碳纳米管透射电镜表征分析
图3为混酸处理前后多壁碳纳米管的透射电镜图。图中显示,未处理碳纳米管管束之间相互缠绕,相互团聚,有明显的颗粒物存在,这不利于掺杂分散。混酸处理后可以明显看出,碳纳米管已经很好地被截断变短,端点突出,管束清晰,颗粒消失,一些含碳物质已被清除,整个物质体系被纯化,分散均匀,这有利于复合材料的制备。同时碳纳米管上引入了更多的羰基和羧基,与红外结果一致,达到了预期设计效果。
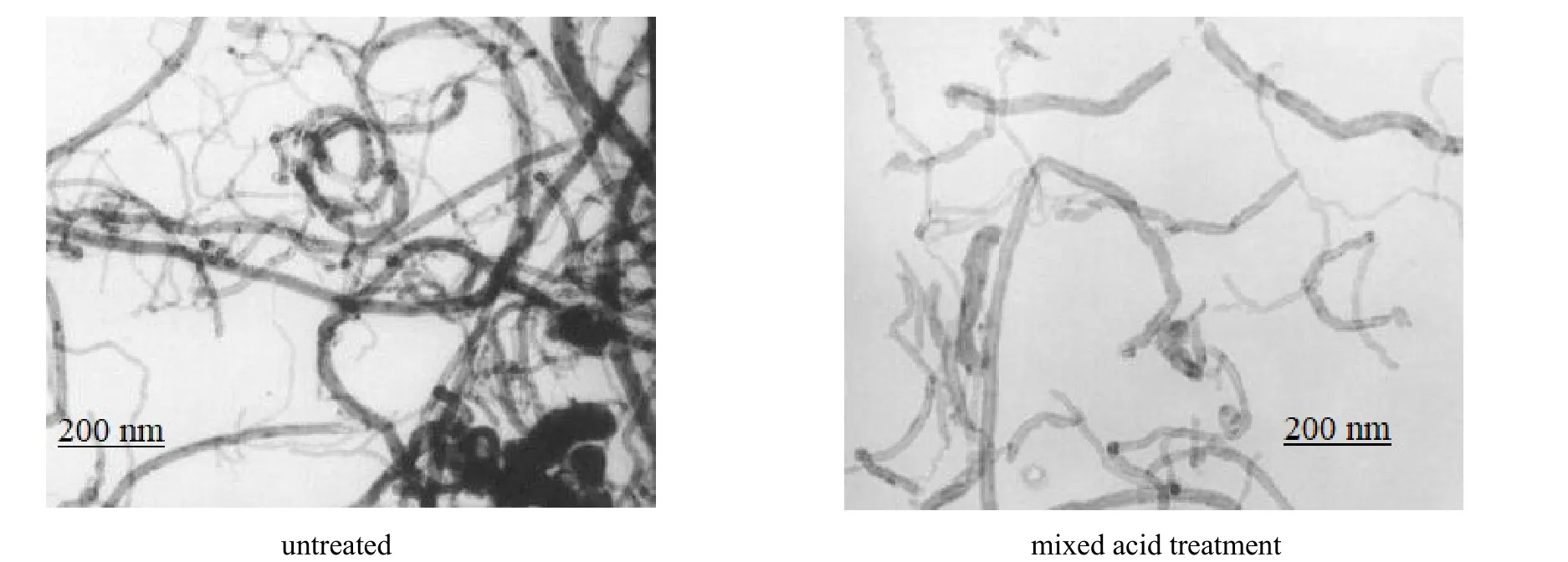
图3 多壁碳纳米管的透射电镜Fig.3 TEM images of multi-walled carbon nanotubes
2.4.3 MWNTs/液晶高分子复合材料的核磁共振氢谱图分析
图4和图5分别为单体4-(噻吩-2-亚甲基氨基)-10-十一烯酸苯酯和原位聚合物核磁共振氢谱。由图4可知,单体氢位移集中在1~3 ppm,主要为烷链烃的氢位移,而图5中氢位移更密集,峰面积更大,表明聚合反应已发生,另外在4.023~4.028 ppm有氢位移,0.873~0.916 ppm也有明显的氢位移,表明原位聚合是多壁碳纳米管且与聚合物有可能产生了氢键,形成了键结合[18-19]。
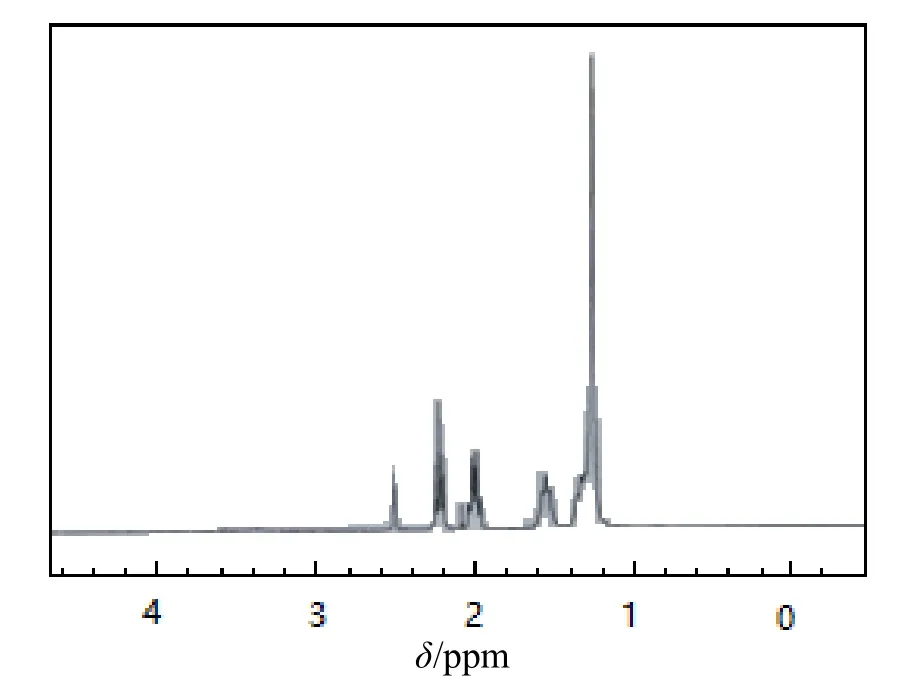
图4 单体4-(噻吩-2-亚甲基氨基)-10-十一烯酸苯酯核磁共振氢谱图谱Fig.4 NMR hydrogen spectra of 4-(thiophene-2-methylamino)-10-undecenoate phenyl ester
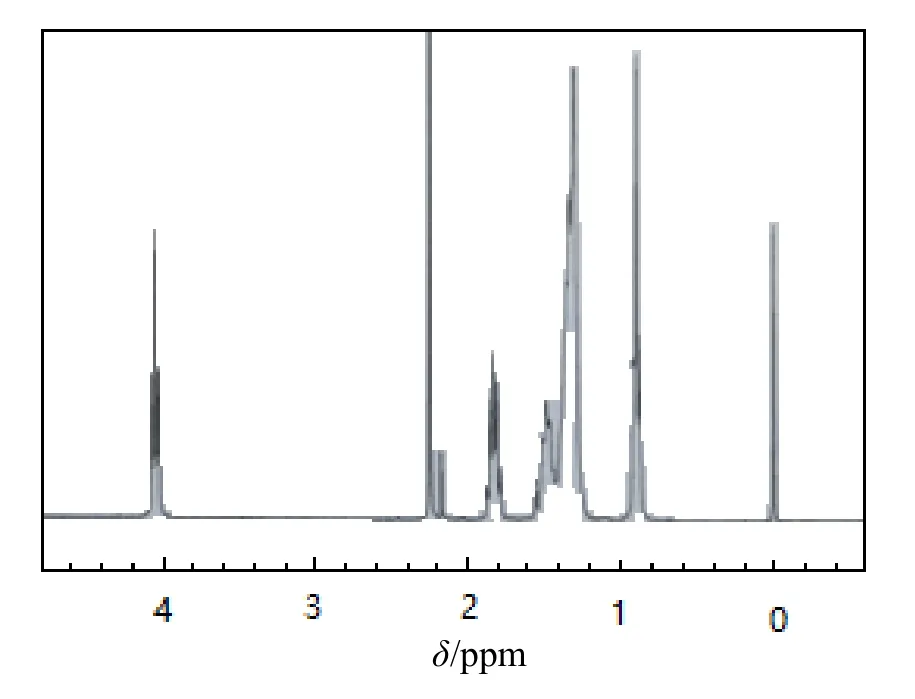
图5 原位聚合物核磁共振氢谱图谱Fig.5 NMR hydrogen spectra of in situ polymers
2.4.4 目标产物的热致偏光显微镜分析
图6和图7为未加改性后多壁碳纳米管在热致偏光显微镜(POM)下的聚合物液晶态和液化态,图8和图9为掺杂结构修饰后的多壁碳纳米管后,热致偏光显微镜下的聚合物液晶态和接近焦化态。由图可以看出,液晶相到液化态液晶相宽只有56 ℃,而掺杂结构修饰后的多壁碳纳米管后,液晶相温度范围增宽,且液晶相条纹状更加明显,即液晶相的涡流线更加明显,并且掺杂的碳纳米管多数排列在条纹处,另外还有一部分排列在液晶相涡流中,该现象表明碳纳米管掺杂有利于形成液晶织构,融化状态,结合依然有序排列,并在升温接近焦化碳化时,其液晶相稳定,优化了材料的性能。

图6 升温到226 ℃POM织构Fig.6 POM texture at 226 ℃

图7 升温到282 ℃POM织构(液化态)Fig.7 POM texture (liquefied state) heated to 282 ℃
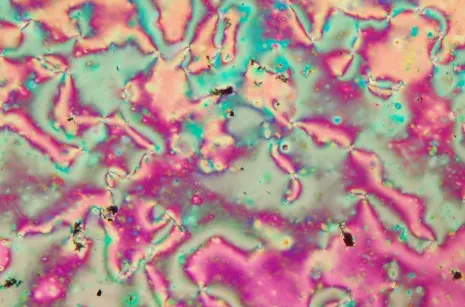
图8 升温到236 ℃POM织构Fig.8 POM texture at 236 ℃

图9 升温到362 ℃POM织构(接近焦化态)Fig.9 POM texture at 362 ℃(near coking state)
3 结 论
采用原位聚合法将制得4-(噻吩-2-亚甲基氨基)-10-十一烯酸苯酯液晶,与经结构修饰后的多壁碳纳米管进行聚合反应。利用微波反应能更有效地达到两者均匀分散、结合、增强界面作用,并且结合稳定,而不是单一的混合,优化了材料的性能及其稳定性。在加热情况下,目标材料熔化,但没有出现两相(基体相与增强相)团簇分离,而是多壁碳纳米管在液晶相涡流线中有序排列,高温作用下仍然保持着较好的液晶相。
因此,利用改性碳纳米管作为柔性高分子液晶材料的中心,在加热熔化时也不会改变它们之间的结合,此类高分子液晶对碳纳米管具有一定的组织排列作用,而碳纳米管对液晶相具有一定的稳定作用,这对液晶复合材料研究提供了较大的应用参考价值。
猜你喜欢
武汉工程大学学报(2022年4期)2022-08-26
建材发展导向(2021年13期)2021-07-28
化工设计通讯(2021年5期)2021-05-26
西北农林科技大学学报(自然科学版)(2021年1期)2021-03-04
浙江农林大学学报(2020年2期)2020-04-22
物理化学学报(2020年1期)2020-04-02
化工设计通讯(2020年8期)2020-01-12
电子制作(2019年19期)2019-11-23
分析化学(2017年12期)2017-12-25
电子制作(2017年13期)2017-12-15
