
基于CRISPR/Cas9 核糖核蛋白体DNA 定点内切酶体外活性建立高效基因型分析技术
2020-07-02 09:47祁显涛刘昌林谢传晓朱金洁
作物学报 2020年7期
王 南 祁显涛,2 刘昌林 谢传晓,* 朱金洁,*
1 中国农业科学院作物科学研究所, 北京100081; 2 安徽农业大学, 安徽合肥230036
2012年9月, Jinek 等[1]在体外验证了sgRNA/Cas9 核糖核蛋白复合体具有DNA 定点剪切的活性。次年, 张峰课题组首次报道了CRISPR/Cas9 在人类细胞中定点编辑DNA 的活性[2]。自此, 基因编辑技术成为生命科学领域的技术发展前沿, 研究热度呈指数级增长。CRISPR/Cas 基因编辑技术不仅可应用于基因敲除、基因插入、定点突变与染色体重组等遗传操作[3-6], 而且还派生出如碱基编辑、转录激活或抑制、表观修饰以及DNA 与RNA 原位示踪成像等一系列重要的遗传学工具[7-11]。然而, 对该系统的体外应用却鲜有报道。基于该系统的DNA 定点剪切的功能, 该系统可进一步开发为位点限制性内切酶,拓展普通限制性内切酶无法靶向的位点, 从而满足日益增长的基因型快速鉴定的需求。
目前, 应用最广泛的为组分优化的 II 型CRISPR/Cas9 系统, 由Cas9 与sgRNA 两个元件组成。在sgRNA 的指导下, Cas9 蛋白可在PAM 基序上游的3 nt 处特异性剪切DNA 双链[12]。为打破Cas9蛋白对PAM 序列的依赖性, 科学家们已开发多种识别不同PAM 序列的Cas9 蛋白变体[13], 其中, 基于蛋白结构优化获得的Cas9NG 的靶向范围最广[14]。目前, 不同版本的CRISPR/Cas9 系统已经广泛应用于不同物种的植物基因组编辑[15-17]。基于经典的CRISPR/Cas9 系统, 我国科学家首次创建了水稻的CRISPR/Cas9 突变体库[18], 我们课题组也针对玉米的ZmWx[19-20]、ZmLg1[21]、ZmMTL[22]等多个位点实现DNA 的定点编辑, 获得了多个具有重要育种价值的突变体材料。如何对基因编辑突变体进行快速的基因型鉴定成为当前该技术发展亟待解决的重要问题。此外, 除基因编辑突变体具有基因型快速鉴定的需求外, 任何基于DNA 序列变异的位点均需要一种快速、经济、可靠、高效与高通量的基因型鉴定技术。例如, 人工诱变及化学诱变群体突变体的快速筛选、基因功能解析中分离群体中个体的基因型快速鉴定、突变体基因型的快速鉴定等, 因此, 建立高效简便的方法对于功能基因组学研究与作物分子育种等均具有重要的应用价值。
目前, 用于植物突变体基因型鉴定的技术主要包括PCR 限制性内切酶酶切(PCR/RE)、错配特异性核酸酶酶切(T7E I/Surveyor assay)、Sanger 测序、二代测序(Next-generation sequencing, NGS)及高分辨率溶解曲线分析(High-resolution melting analysis,HRMA)等, 这些技术都存在自身的优点, 但同时也有局限性。PCR/RE 技术严重依赖于突变碱基附近的限制性内切酶识别位点, 限制了靶点设计的自由度从而限制了其范围[23]。基于DNA 双链错配原理的T7E I 或CEL I 酶切检测, 商业酶价格高昂, 技术上也存在不能有效区分纯合突变与野生型以及杂合突变与双等位基因突变的限制[24]。Sanger 测序虽可直观地呈现靶点突变信息, 但其价格昂贵, 不适用于大规模突变群体检测。NGS 及衍生的Hi-TOM 技术主要适用于高通量筛选鉴定, 无法满足对小量与中量样本突变体材料早期快速鉴定的需要[25]。2018年高彩霞团队首先尝试开发了基于 CRISPR-Cas9系统的PCR/RNP 技术[26], 该技术不受限制性酶切位点选择的局限, 同时可精准区分野生型、纯合突变及杂合突变等基因型, 成本低廉、快速高效, 可实现突变体高通量筛选, 但检测体系仍需要优化,进一步提高检测效率、特异性与灵敏度, 才能进一步推广应用。
本研究的目标是建立与优化基于CRISPR/Cas9与CRISPR/Cas9NG 核糖核蛋白体定点DNA 内切酶体外活性的基因型高效分析技术, 为该技术的高效应用奠定完善的技术方案。本研究利用原核表达并纯化的Cas9 蛋白或Cas9NG 蛋白为DNA 内切酶, 以体外转录的靶向ZmWx基因靶点的 sgRNA 或esgRNA 为向导 RNA 分子, 通过体外组装为sgRNA/Cas9-RNP 复合体, 对实验室通过基因编辑手段获得的玉米ZmWx突变体靶点PCR 产物进行酶切, 从而实现对突变体的基因型分析。本研究将为突变体的高效基因型分析提供技术参考。
1 材料与方法
1.1 材料
基于ZC01 玉米自交系背景创制的靶向ZmWx基因CRISPR-Cas9 基因编辑等待基因型分析的玉米材料[19], BL21(DE3)大肠杆菌表达感受态(北京全式金生物技术有限公司, 货号3CD801), PET30a-Cas9 载体(构建), PET 30a-Cas9NG载体(构建)。
1.2 方法
1.2.1 Cas9 及Cas9NG 蛋白的原核表达载体构建
从本实验室保存载体上设计引物(表1), 扩增获得Cas9 片段, 使用限制性内切酶NcoI 和NheI 双酶切PET 30a 载体, 琼脂糖凝胶电泳分离后, 进行胶回收纯化。回收纯化的Cas9 片段与PET 30a 线性载体, 用NEB 组装酶(北京NEB 公司) 50 ℃, 1 h进行组装, 转化至BL21(DE3)大肠杆菌表达感受态中, 均匀涂布在卡那抗性的LB 固体培养基上, 37℃过夜培养, 挑取单克隆酶切鉴定, 将获得的阳性克隆质粒送上海英潍捷基公司测序。测序结果与Snapgene 载体图谱比对一致, 即获得PET 30a-Cas9 载体。用上述方法, 将限制性内切酶NheI 替换为Hind III, 构建得到PET 30a-Cas9NG 载体。
1.2.2 Cas9 及Cas9NG蛋白的原核表达与纯化
将构建好的融合His标签的Cas9 和Cas9NG蛋白原核表达载体转化到原核表达菌株BL21(DE3)中。挑取单克隆, 2 次扩大培养至OD600在0.7~0.8 之间。冰浴后加入0.5 mol L-1IPTG溶液诱导蛋白表达。以下步骤均在4℃进行: 超声破碎裂解后, 取上清, 进行Ni-NTA琼脂糖介质亲和层析, 然后进行阳离子交换柱层析, 收集各组分进行10% SDS-PAGE鉴定, 根据凝胶电泳结果收集含有洗脱蛋白的各组分, 最后,用100,000-MWCO蛋白超滤管将含有洗脱蛋白的组分缓冲液替换为蛋白存储液(50 mmol L-1Hepes,150 mmol L-1KCl, 1 mmol L-1TCEP, 20% Glycerol,pH 7.5)。分装后液氮速冻, 于−80°C冰箱保存备用。
1.2.3 Cas9 及Cas9NG蛋白的定量 对待测样品进行BSA (bovine serum albumin)粗定量后, 使用BCA蛋白快速测定试剂盒进行蛋白精确定量(上海英潍捷基公司), 首先制备蛋白标准品梯度稀释液和BCA工作液, 然后分别取标准品和待测样品20 μL加入到微孔板中, 之后加入20 μL工作试剂, 充分混匀, 震荡30 s, 室温静置5 min, 用分光光度计测量蛋白标准品和待测样品在480 nm处的吸光度。最后作出蛋白标准品吸光度相对于浓度的标准曲线, 根据待测样品在480 nm处的吸光度, 在标准曲线上查出待测样品相当于标准蛋白的量, 从而计算得出待测样品的浓度。
1.2.4 RNA体外转录 利用引物重叠PCR原理,设计引物sgRNA scaffold F、sgRNA scaffold R (表1)扩增获得sgRNA转录模板, 用2%琼脂糖凝胶电泳分离后, 回收纯化。使用T7 转录试剂盒(北京NEB公司)进行体外转录, 体外转录体系为2 μL模板, 15 μL NTP Mix, 3 μL T7 转录酶, RNase-free H2O补至40 μL。37℃孵育3.5 h。结束后, 加入2 μL DNase I、8 μL RNase-free H2O消化模板DNA。然后用RNA纯化浓缩试剂盒(ZYM公司)进行纯化浓缩, 分光光度计测得浓度后, 通过10% Urea-PAGE鉴定sgRNA的质量。液氮冷冻, −80℃保存备用。采用相同方法获得高纯度esgRNA。
1.2.5ZmWx突变体DNA提取 切取玉米ZmWx突变体种子部分, 充分研磨, 置于1.5 mL EP管中,加入1 mL裂解缓冲液(10 mmol L-1Tris-HCl, 1 mmol L-1EDTA, pH 8.0, 0.1% SDS, pH 8.0), 混匀后于65℃水浴1 h, 10,000×g离心2 min, 小心吸取上清液于一支新的1.5 mL EP管中, 加入等体积的酚/氯仿/异戊醇(25∶24﹕1)(北京酷来博生物技术有限公司),充分混匀后, 10,000×g离心10 min, 吸取上清液于一支新的1.5 mL EP管中, 加入0.2 倍体积7.5 mol L-1的醋酸铵和2 倍体积的无水乙醇沉淀DNA, 10,000×g离心1 min, 弃上清液, 用70%无水乙醇洗涤2 次,弃上清液, 室温干燥, 用0.2 mL TE溶液溶解DNA,−20℃保存备用。
1.2.6 PCR/RNP酶切 以种子DNA为模板, 设计引物ZmWxF、ZmWxR (表1), 通过PCR扩增sgRNA靶标两侧各300~500 bp序列。PCR程序为95℃预变性3 min; 94℃变性25 s, 63℃退火25 s, 72℃延伸30 s,35 个循环; 72℃总延伸2 min; 4℃保存。将X μg Cas蛋白、X μg esgRNA、2 μL Buf 3.1 (北京NEB公司)和(12-X) μL RNase-free H2O混合均匀, 37℃孵育0.5 h进行预组装, 加入6 μL PCR产物, 37℃孵育2 h。然后加入2 μL RNaseA, 37℃孵育15 min以消化RNA, 65℃孵育20 min使蛋白质变性, 终止反应。结束后进行2%琼脂糖凝胶电泳检测。将ZmWx突变体PCR产物送上海英潍捷基公司测序, 以验证酶切检测结果。
1.2.7 引物设计 本研究所用引物除ZmWxF、ZmWxR由Primer premier软件设计外, 其他引物均由Snapgene软件设计, 并委托上海英潍捷基公司合成。引物信息见表1。
2 结果与分析
2.1 CRISPR-Cas9、Cas9NG 蛋白原核表达与纯化
构建Cas9 与Cas9NG 蛋白原核表达载体, 载体的主要元件有T7 启动子、Lac 操纵子、His 蛋白纯化标签、目标蛋白的CDS 序列及T7 终止子等(图1-A)。通过亲和纯化与阳离子交换纯化两步法对原核表达的Cas9 与Cas9NG 蛋白纯化后, 获得纯度较高的Cas9 与Cas9NG 蛋白制品(图1-B, C)。结果表明, 收集1 L 诱导表达的大肠杆菌可纯化约4 mg的Cas9 蛋白, 且经过阳离子交换柱二次纯化获得的蛋白样品, 纯度显著提高。经过一次原核表达及纯化所获得的蛋白制品在产量和纯度上可满足后续酶切实验需要。
2.2 RNA 的体外转录与基于esgRNA/Cas9-RNP和esgRNA/Cas9NG-RNP 检测体系的初建立
目前, 应用最为广泛的sgRNA 骨架序列为81 nt(图2-A), 后续研究发现将sgRNA 骨架序列的poly U位点的第4 个碱基突变为C 以及延长RNA 双链部分5 个碱基对(图2-B), 可有效提高基因编辑的效率[27-28]。因此, 本研究采用两种sgRNA 骨架序列进行Cas9-RNP 与Cas9NG-RNP 实验体系的建立。首先, 设计体外转录靶向本实验室已发表ZmWx基因编辑位点的sgRNA 或esgRNA 引物, 见表1 所示。通过PCR 扩增获得体外转录的DNA 模板后, 利用T7 转录酶进行体外RNA 转录, 转录产物经过纯化后, 经10% Urea-PAGE 电泳鉴定。结果显示, 经过体外转录的sgRNA 条带大小在100 nt 左右, esgRNA条带大小在110 nt 左右, 与预期RNA 条带大小一致且质量较好, 纯化后esgRNA 有部分杂带(图2-C),推测可能是由体外转录DNA 模板回收不纯所致。

表1 本研究所用引物Table 1 Primer sequences used in this paper
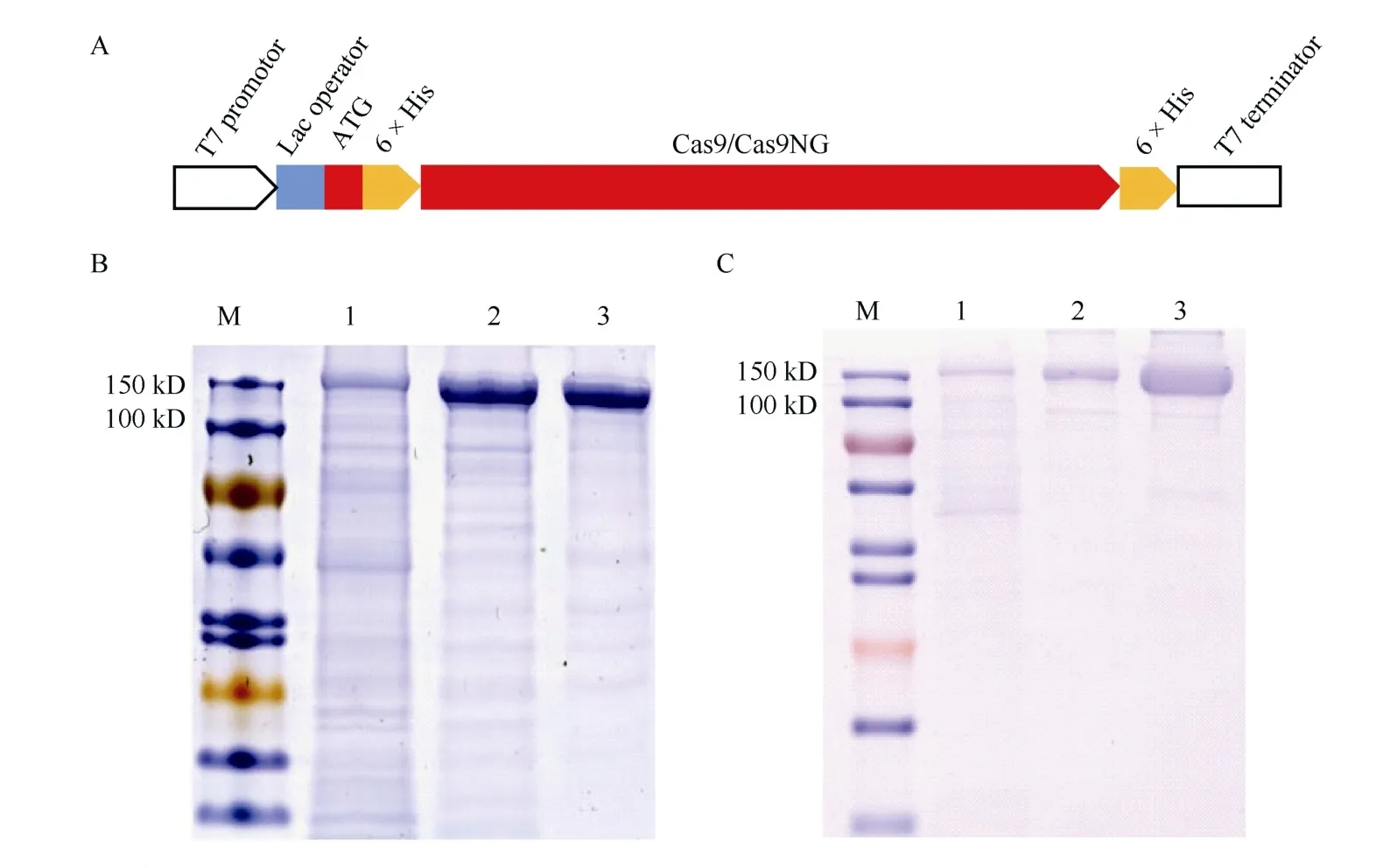
图1 Cas9 与Cas9NG 蛋白的原核表达及纯化Fig. 1 Purification of Cas9 and Cas9NG protein

图2 sgRNA、esgRNA 体外转录及基于esgRNA/Cas9-RNP 和esgRNA/Cas9NG-RNP 检测体系的建立Fig. 2 In vitro transcription of sgRNA and esgRNA, as well as the establishment of cleavage assay via esgRNA/Cas9-RNP and esgRNA/Cas9NG-RNP
以野生型ZmWx种子DNA 为模板, 通过PCR扩增sgRNA 靶标两侧各300~500 bp 序列, 作为酶切底物。然后将 Cas9 蛋白、Cas9NG 蛋白分别与sgRNA、esgRNA 预组装为4 种RNP 复合体, 对PCR产物进行酶切, 结束后进行琼脂糖凝胶电泳检测,以验证RNP 活性。结果显示, sgRNA/Cas9-RNP 或sgRNA/Cas9NG-RNP 在ZmWx位点上不具有酶切活性, 底物 DNA 均没有被切开。而基于 esgRNA/Cas9-RNP 或基于esgRNA/Cas9NG-RNP 可对ZmWx靶点底物DNA 进行充分酶切, 酶切产生400 bp 与500 bp 两条DNA 条带(图2-D)。
2.3 基于esgRNA/Cas9-RNP 和esgRNA/Cas9NGRNP 检测体系的优化及其对不同基因型的区分
针对esgRNA/Cas9-RNP 或esgRNA/Cas9NGRNP 两种突变体基因型检测方法进行酶切体系的进一步优化。采用控制变量法分别研究了esgRNA 用量、Cas9 或Cas9NG 蛋白用量和酶切反应时间等对酶切效率的影响。对于 esgRNA/Cas9-RNP, 当esgRNA 和Cas9 蛋白均为1 μg 时, 酶切反应时间为0.5 h, 可实现对500 ng 靶位点PCR 扩增产物的充分酶切(图3-A~C)。对于esgRNA/Cas9NG-RNP,当esgRNA 和Cas9NG 均为2 μg 时, 酶切反应时间为4 h, 实现对500 ng 靶位点PCR 扩增产物的充分酶切(图 3-E~G)。采用优化后的 esgRNA/Cas9-RNP 与esgRNA/Cas9NG-RNP 检测体系, 分别对ZmWx基因编辑产生的3 种不同基因型材料进行基因型鉴定, 以验证基于 sgRNA/Cas9-RNP系统是否能够准确区分不同的基因型。优化后的esgRNA/Cas9-RNP 与esgRNA/Cas9NG-RNP 检测体系均具备准确区分不同基因型PCR 产物的能力,能够有效区分野生型、纯合突变体及杂合突变体材料(图3-D, H)。
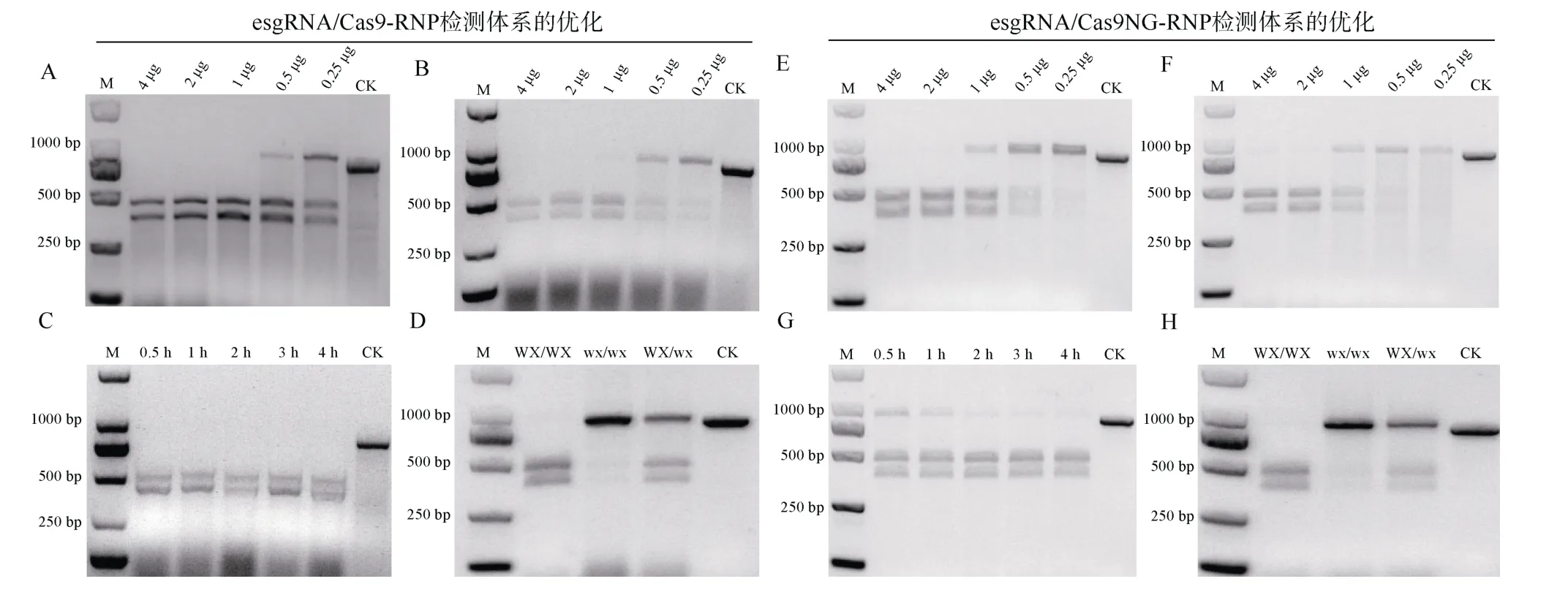
图3 基于esgRNA /Cas9-RNP 及esgRNA/ Cas9NG-RNP 检测体系的优化Fig. 3 Optimization of cleavage assay via esgRNA /Cas9-RNP and esgRNA/ Cas9NG-RNP
2.4 利用Cas9NG 拓宽检测位点范围
前人的研究结果表明, Cas9NG 作为spCas9 的变体, 由于替换了PI 功能域中(PAM-interacting, PI)识别PAM 的第2 和第3 位碱基的氨基酸, 使其不再具有识别特异性, 从而可以识别PAM 为“NG”基序, 极大地拓展了基因编辑的范围[13-14]。因此, 我们探索应用Cas9NG 拓宽检测位点范围。
我们在ZmWx编辑位点附近重新设计了6个PAM为“NG”的靶位点(图4-A), 通过体外转录及凝胶电泳检测发现, 6 个esgRNA的分子量大小正确, 转录效果较好(图4-B)。应用优化后的esgRNA/Cas9NG-RNP体系, 将Cas9NG与6 种esgRNA分别组装成RNP, 对野生型ZmWx靶位点PCR扩增产物进行酶切。从图4-C可以看出, 仅有第6 组对野生型WaxyPCR 产物有酶切活性, 而第 6 组esgRNA 的PAM 为“AGG” 。 结 果 表 明, 利 用esgRNA/Cas9NG-RNP并不能拓宽ZmWx靶位点检测的范围。
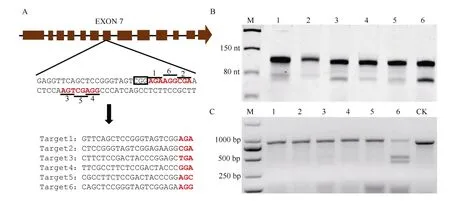
图4 基于esgRNA/Cas9NG-RNP 检测体系对不同ZmWx 位点的检测Fig. 4 Cleavage assay of different targets for ZmWx locus via esgRNA/Cas9NG-RNP
2.5 基于esgRNA /Cas9-RNP 检测体系筛选基因编辑突变体
由于esgRNA/Cas9-RNP在检测反应中RNP用量、酶切反应时间及剪切效率均优于esgRNA/Cas9NGRNP, 本研究利用esgRNA/Cas9-RNP系统对ZmWx基因编辑突变群体进行基因型检测。结果表明, 对随机选择的32 个ZmWx基因编辑材料的靶位点进行酶切后,ZmWx野生型、纯合突变体及杂合突变体均得到有效区分(图5-A)。为进一步确定利用esgRNA/ Cas9-RNP进行突变体基因型鉴定的可靠性, 我们随机选择3 种基因型的一个样品进行Sanger测序。通过峰图判读与序列比对可知, 测序结果与检测结果一致(图5-B)。
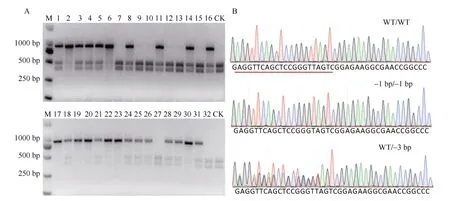
图5 基于esgRNA/Cas9-RNP 的ZmWx 基因编辑突变体检测Fig. 5 Genotyping genome-edited ZmWx mutant via esgRNA/Cas9-RNP
3 讨论
建立一种快速、经济、高效与高通量的突变体基因型鉴定技术对作物基因功能鉴定等基础研究与分子育种研究等方面具有广泛的应用价值。因此,此类技术具有重要研发价值。此前已研发了多种基因型鉴定方法, 本方法与其相比, 在应用范围与简便高效性上具有重要的优势。以当前应用较广的基于限制性内切酶的PCR/RE 检测和基于DNA 双链错配原理的T7E I 或CEL I 酶切检测为例。PCR/RE 方法严重依赖于突变位点处具有合适的II 型限制性内切酶位点, 识别位点为6 bp 的限制性内切酶约4K DNA 区间才能有一个合适的位点, 而依赖CRISPR/Cas9 PAM-NGG 基序的特征, 约16 bp 即能有一个合适的位点, 约为前者的260 倍。基于DNA 双链错配原理的T7E I 或CEL I 酶切检测无法有效区分纯合突变体与野生型, 双等位基因突变体和杂合突变体,商业化的酶价格高昂, 检测体系还依赖PAGE 胶,成本高昂, 实验体系操作繁琐。而本研究采用的方法, 经过一次原核表达及纯化获得的Cas9 蛋白(4 L原核表达菌液)可用于至少10,000 个酶切体系的检测, 不同位点突变体材料的检测只需替换与靶位点匹配的sgRNA 即可, 体外转录1 个反应的esgRNA可用于至少200 个酶切体系检测, 合计1 个反应的成本在1 元以下。整个检测反应可在1~3 h 内完成,检测都是基于简易的琼脂糖凝胶分析, 能够满足对突变体进行早期高通量基因型的快速高效分析。此外, 基于sgRNA/Cas9-RNP 的检测体系还具有应用范围广的特点, 不仅适用于由 ZFN、TALEN、CRISPR-Cas9、CRISPR-Cpf1 等序列特异性核酸酶定点编辑突变体的基因型分析, 还适用于自然变异、人工诱变突变体在特定位点上的基因型分析。
我们在本实验积累的经验还表明,ZmWx位点对应的sgRNA 无法介导Cas 蛋白活性, 推测原因是通过T7 启动子体外转录靶向ZmWx位点的sgRNA, 在体外酶切缓冲液中不能形成稳定的二级结构, 从而不能与Cas 蛋白形成有剪切活性的核糖核蛋白体(RNP)。因此, 将 sgRNA 替换为骨架序列优化的esgRNA 是必要的, 该优化会显著提高检测体系对位点的兼容性, 优化的 esgRNA 分别与 Cas9 及Cas9NG 蛋白组装形成的RNP 复合体可实现底物DNA 的充分酶切, 这与前人研究的结论是一致的,即通过对sgRNA 骨架序列优化, 可以提高sgRNA结合靶位点的能力, 从而提高CRISPR-Cas9 的切割效率[27-28]。此外, 本研究发现, 基于 esgRNA/Cas9NG-RNP 的检测体系, 除在PAM 为“NGG”序列的6 号靶位点有较强的剪切活性外, 在PAM 为非“NGG”的靶位点均没有检测到明显的剪切活性。推测原因可能是Cas9NG 蛋白变体对于靶向位点具有一定的偏好性, 不同的靶位点由于GC 含量的不同、碱基的分布差异, sgRNA 识别结合靶位点的能力有所不同, 基于esgRNA/Cas9NG-RNP 的检测体系还有待于蛋白活性的进一步优化。该数据对利用Cas9NG 的NG-PAM 基序特征扩大体内CRISPR/Cas9 基因编辑设计位点自由度具有重要参考价值。
4 结论
利用Cas9 或Cas9NG 变体蛋白与sgRNA 核糖核蛋白复合体(sgRNA/Cas9-RNP 或 sgRNA/Cas9NG-RNP)体外DNA 定点内切酶活性, 建立并优化了一种简便、高效与低成本的基因型分析技术。esgRNA/Cas9 形成的 RNP 复合体核酸酶活性比esgRNA/Cas9NG 高, 酶切等量底物需要的RNP 复合体的量少且时间更短, 可用于突变体基因型的高通量鉴定。利用esgRNA/Cas9NG 拓宽目标靶位点的检测数据为CRISPR/Cas9NG 活体基因编辑技术研发提供了重要参考数据。
猜你喜欢
传染病信息(2022年4期)2022-11-23
西部医学(2022年9期)2022-09-26
亚热带农业研究(2022年1期)2022-08-08
中国农学通报(2022年12期)2022-06-01
中国糖料(2022年2期)2022-04-06
中国种业(2021年11期)2021-11-25
教学考试(高考生物)(2021年2期)2021-05-31
农业科技通讯(2021年1期)2021-03-06
中国农业科技导报(2020年3期)2020-03-15
山西农业科学(2020年2期)2020-02-29
