
基于密码子偏性对应分析数据的牛病毒性腹泻病毒分型研究
2020-07-01 13:06马小静张巧娥李继东
甘肃农业大学学报 2020年2期
马小静,张巧娥,李继东
(宁夏大学农学院,宁夏 银川 750021)
牛病毒性腹泻病毒(bovine viral diarrhea virus,BVDV)是一种与牛胃肠道、呼吸道和生殖道疾病相关的重要病原体,其所导致的繁殖机能下降给全球养牛业造成了持续的经济损失.妊娠母牛感染BVDV后会出现流产,产出畸形胎儿或木乃伊胎,甚至产出持续感染牛(PI牛),而PI牛是BVDV最主要的传染源[1].BVDV是一种有囊膜的单股正链线性RNA病毒,属于黄病毒科瘟病毒属,其基因组长约 12.5 kb,由5′非翻译区(5′untranslated region,5′UTR)、一个大的开放性阅读框(open reading frame,ORF)和3′非翻译区(3′untranslated region,3′UTR)构成.ORF编码3 988个氨基酸,由细胞和病毒蛋白酶加工后至少生成 12种成熟蛋白质,包括Npro,C,ERNS,E1,E2,p7,NS2,NS3,NS4A,NS4B,NS5A,NS5B[2-3].Npro是一种非结构蛋白,它能够产生自己的C端.核衣壳蛋白C和3种包膜糖蛋白ERNS、E1、E2代表BVDV的结构蛋白.E2是瘟病毒中高度可变的、具有免疫学优势的糖蛋白,是中和抗体的主要靶标[4].5′-UTR是病毒基因组中高度保守的部分,其包含一个内部核糖体进入位点,与病毒多种蛋白的翻译有关.
目前,瘟病毒属包括4种已被国际病毒分类委员会(ICTV)认可批准的病毒:BVDV-1,BVDV-2,经典猪瘟病毒(CSFV)和绵羊边界病病毒(BDV).此外,Schirrmeier等从巴西源的胎牛血清中分离到Hobi样病毒,即BVDV-3型[5],在对其进行遗传学和抗原学分析后,将其归为黄病毒科瘟病毒属,之后也陆续从意大利、巴西等地的自然病例中分离得到.基因组的不同区域,如5′UTR、Npro、E2、NS2-3和NS5B等已被用于BVDV和其他瘟病毒的基因分型和分类,如构建分子进化树常用NS5B内的RNA聚合酶(RdRP)基因和5′UTR序列.根据基因组5′UTR的差异,将BVDV分为2种基因型:BVDV-1型和BVDV-2型[6].5′UTR序列最常用于BVDV分离株的系统发育分析和基因分型,其次是Npro和E2基因序列.迄今为止,几乎所有BVDV亚基因型都根据这3个基因组区域进行分类.对BVDV分离株的Npro和E2基因序列进行分析,能够更可靠的将其分配到已建立的和新鉴定的亚基因型中.自20世纪90年代初期鉴定了2种BVDV-1亚基因型之后,至今已分离到至少22种BVDV-1亚基因型(BVDV1a-1v).依据BVDV-2型5′端非编码区存在的二级结构差异性,Giangasper M将其进一步细分为4个亚型,BVDV2a、BVDV2b、BVDV2c和BVDV2d[7].Vilcek关于BVDV基因分型结果表明,亚基因型的分布与病毒分离株的地理来源无关[8].2007年,任敏从新疆等地的病料中首次分离到了BVDV-2型的病毒,这是我国首次报道关于BVDV-2型毒株的分离与鉴定[9].根据体外细胞培养特性,BVDV-1型和BVDV-2型又进一步被细分为细胞致病型(cp)和非细胞致病型(ncp).值得一提的是,ncp BVDV对胎儿先天免疫应答的调节和建立BVDV持续感染具有重要意义[10].
密码子偏好性(condon usage bias,CUB)是指同义密码子在编码氨基酸时使用频率并不完全相同的现象[11].目前,一些参数已被广泛用于研究密码子偏好性,包括相对同义密码子使用度(relative synonymous codon usage,RSCU),密码子适应指数(codon adaptation index,CAI)以及最优密码子使用频率(frequency of optimal codons,FOP)等.2010年,Fu等[12]对疱疹病毒的密码子偏好性进行研究后指出,Tupaiid疱疹病毒Ⅰ型(TuHV-1,Tupaiid herpesvirus 1)的密码子偏好性和巨细胞病毒相似,且基于氨基酸序列或完整基因组序列的系统发育分析结果也显示这2种病毒关系密切.此外,一项针对于羊痘病毒密码子的研究发现,系统发育上彼此接近的病毒,特别是同一属的成员,在其编码区共享相似的核苷酸组成模式并采用几乎完全相同的最优密码子[13].以上研究发现都为密码子偏好性在病原分型中的应用提供了更多的证据.
基于以上研究结果,本研究对数据库中BVDV基因组序列进行分析,计算密码子偏好性参数,对所得数据进行对应分析,进一步对分析所得数据进行聚类,聚类结果和BVDV分子进化树进行比对,探索基于密码子偏性的病毒分型方法.
1 材料与方法
1.1 数据来源
本文数据来源于NCBI的核酸数据库Nucleotide,检索到2019年1月为止所有BVDV基因组序列和BDV、CFSV基因组参考序列,包括BVDV-1、BVDV-2、BVDV-3共计104条核酸序列.具体信息见表1.

表1 BVDV序列信息
续表1 Continuedtable1检索号Accession病毒株Virusstrain国家CountryMH166806BVDV-1/XC中国ChinaMH379638BVDV-1/Ho916英国UKMH490942BVDV-1/BJ-2013中国ChinaMH490943BVDV-1/BJ-2016中国ChinaAB567658BVDV-2/Hokudai-Lab/09日本JapanAB894423BVDV-2/GBK_E+日本JapanAB894424BVDV-2/GBK_E-日本JapanAF002227BVDV-2/C413美国USAAF502399BVDV-2-NewYork'93德国GermanyFJ527854BVDV-2/XJ-04中国ChinaGQ888686BVDV-2/JZ05-1中国ChinaHG426479BVDV-2/D37-13-2_Dup(-)德国GermanyHG426480BVDV-2/D37-13-2_Dup(+)德国GermanyHG426481BVDV-2/D75-13-609_Dup(-)德国GermanyHG426482BVDV-2/D75-13-609_Dup(+)德国GermanyHG426483BVDV-2/NRW12-13_Dup(-)德国GermanyHG426484BVDV-2/NRW12-13_Dup(+)德国GermanyHG426485BVDV-2/NRW14-13_Dup(-)德国GermanyHG426486BVDV-2/NRW14-13_Dup(+)德国GermanyHG426487BVDV-2/NRW19-13-1_Dup(-)德国GermanyHG426488BVDV-2/NRW19-13-1_Dup(+)德国GermanyHG426489BVDV-2/NRW19-13-8_Dup(-)德国GermanyHG426490BVDV-2/NRW19-13-8_Dup(+)德国GermanyHG426491BVDV-2/Potsdam1600德国GermanyHG426492BVDV-2/SH2210-14德国GermanyHG426493BVDV-2/SH2210-17德国GermanyHG426494BVDV-2/SH2210-23德国GermanyHG426495BVDV-2/VOE4407德国GermanyHQ258810BVDV-2/SH-28中国ChinaJF714967BVDV-2/HLJ-10中国ChinaKC963968BVDV-2/11F011韩国KoreaKJ000672BVDV-2/SD1301中国ChinaKT832817BVDV-2/USMARC-60764美国USAKT832818BVDV-2/USMARC-60765美国USAKT832819BVDV-2/USMARC-60766美国USAKT832820BVDV-2/USMARC-60767美国USAKT832821BVDV-2/USMARC-60768美国USAKT832822BVDV-2/USMARC-60779美国USAKT832823BVDV-2/USMARC-60780美国USAKX096718BVDV-2/HB-1511中国ChinaLC006970BVDV-2/KZ-91-CP日本JapanMG879027BVDV-2/CN10.2015.821意大利ItalyMH806434BVDV-2/125c美国USAMH806435BVDV-2/1336H美国USAMH806436BVDV-2/296c美国USAMH806437BVDV-2/9231美国USAMH806438BVDV-2/McCart_c美国USAAB871953BVDV-3/D32/00_'HoBi'日本JapanFJ040215BVDV-3/Th/04_KhonKaen泰国Thailand

续表1 Continuedtable1检索号Accession病毒株Virusstrain国家CountryHQ231763BVDV-3/Italy-1/10-1意大利ItalyJQ612704BVDV-3/Italy-83/10-ncp意大利ItalyJQ612705BVDV-3/Italy-83/10-cp意大利ItalyJX469119BVDV-3/JS12/01中国ChinaJX985409BVDV-3/CH-KaHo/cont瑞典SwedenKC297709BVDV-3/LVRI/cont-1中国ChinaKC788748BVDV-3/Italy-129/07意大利ItalyKJ627179BVDV-3/Italy-68/13ncp意大利ItalyKJ627180BVDV-3/Italy-68/13cp意大利ItalyKU563155BVDV-3/HN1507中国ChinaKY683847BVDV-3/SV757/15巴西BrazilKY762287BVDV-3/PB22487巴西BrazilKY767958BVDV-3/SV478/07巴西BrazilAF037405BDV/X818德国GermanyAF326963CSFV/Eystrup瑞士Switzerland
1.2 方法
1.2.1 构建BVDV分子进化树 检索并整理BVDV及BDV、CFSV的RNA聚合酶(RdRP)基因序列以及5′UTR序列,用生物信息学软件MEGA中的N-J法构建分子进化树.
1.2.2 计算、分析BVDV密码子数据 用软件CodonW中的Correspondence analysis功能模块,对RSCU、密码子使用(codon usage)和氨基酸使用(amino acid usage)进行对应分析(correspondence analysis,COA),其原理和计算方法如下:
在对基因密码子使用概率分析时,将每1条基因作为1个对象,相对密码子使用度作为变量,采用59个同义密码子[去除编码蛋氨酸(M)的密码子AUG和编码色氨酸(W)的密码子UGG以及3个终止密码子]的RSCU值对其密码子使用偏性进行分析,基因间的距离规定为同义密码子相对使用度的欧拉平方距离.对于基因a与基因b,其密码子使用距离的计算公式为:
计算完成后,用Excel对RSCU、codon usage和amino acid usage的COA数据作图,以Axis1为横坐标、Axis2为纵坐标,分析数据特征[12-13].
1.2.3 密码子统计数据的聚类分析 运用统计软件Statistica 12分析上述BVDV密码子对应分析数据.用Cluster Analysis中的Joining(Tree Clustering)进行聚类分析,参数设置为Amalgamation(linkage)rule:Single Linkage;Distance measure:Euclidean distances
2 结果与分析
2.1 分子进化树构建结果
基于BVDV、BDV、CFSV病毒RdRP基因的分子进化树显示,BVDV-1、BVDV-2、BVDV-3分别形成了3个大的分支;在以BDV、CFSV为外群的情况下,BVDV-1和BVDV-2亲缘关系最近,BVDV-3与二者关系稍远;各型的病毒株都处于相应的分支上,部分来自同一地区或国家的分离株都聚在同一较小分支上,表明这些分离株关系最近(图1-A),如分离自德国的BVDV-2/D37-13-2_Dup(-)等12株BVDV-2型毒株和分离自意大利的BVDV-3/Italy-1/10-1等6株BVDV-3型毒株都聚在同一个小分支上;但BVDV-1型的分离株没有典型的地区近缘性.基于5′UTR序列的分子进化树结果与RdRP基因的分析结果类似,3型BVDV分属于不同分支,但在各型内部不同病毒株的分支关系上,两者存在一定差距(图1-B),分离自德国的BVDV-2/D37-13-2_Dup(-)等12株BVDV-2型毒株间的关系和基于RdRP基因分析的结果则表现一致.
2.2 BVDV密码子COA数据分析结果
CodonW计算所得BVDV密码子使用对应分析(COA)数据分析显示,不同类型的BVDV在RSCU、密码子使用和氨基酸使用方面具有型特异性(图2).BVDV-1、BVDV-2、BVDV-3在RSCU和密码子使用上的COA数据分布模式高度相似,3个基因型的分布相对集中于不同象限中,其中BVDV-3基因型全部聚于第一象限.BVDV-1的数据主要分布于第四象限,BVDV-2则主要位于第二象限,两型数据有一定离散,但无交叉分布(图2-A~B).在氨基酸使用方面的数据分布情况类似于RSCU和密码子使用,但所属象限不同,BVDV-3全部位于第三象限,BVDV-1主要位于第二象限,BVDV-2则主要位于第四象限.以上结果表明不同基因型BVDV在RSCU、密码子使用和氨基酸使用方面存在差异,且相对于其他2种基因型来说,BVDV-2和CSFV、BDV的COA数据分布更为接近.

A:基于RdRP的进化树;B:基于5′UTR的进化树.
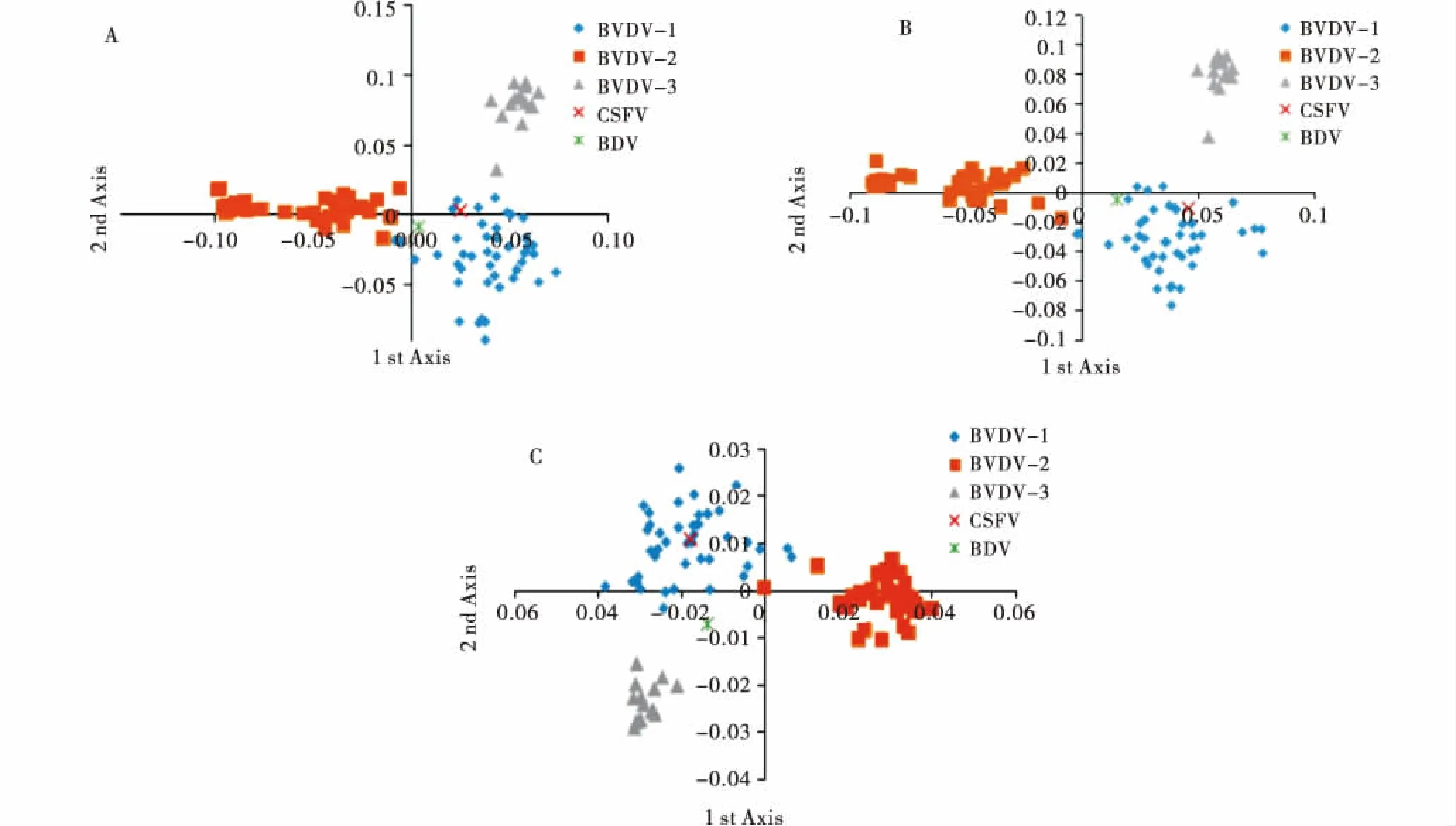
A:相对同义密码子使用度对应分析;B:密码子使用对应分析;C:氨基酸使用对应分析.
2.3 BVDV密码子数据聚类分析结果
RSCU、ENC对应分析数据的聚类分析结果显示(图3),BVDV-1、BVDV-2、BVDV-3分别聚集形成一个大类,和基于RdRP基因以及5′UTR序列的分子进化树有高度统一性;CSFV、BDV和BVDV-1、BVDV-2关系较近,BDV和BVDV-1、BVDV-2关系则更近;来自同一地区的分离株聚在一起,基本处于同一个分支上.
由BVDV-2型病毒株的聚类分析和分子进化树结果可见,发现于德国且分离于同一宿主的BVDV-2/D37-13-2_Dup(-),BVDV-2/D75-13-609_Dup(-),BVDV-2/NRW 12-13_Dup(-)等12株病毒株聚为一类,不仅说明其亲缘关系高度接近,还提示在密码子使用偏好性上这些病毒株也具有高度相似性.来自美国的BVDV-2/USMARC-60764,BVDV-2/USMARC-60765等6株病毒分离株也处于同一较大分支上,表示其在亲缘关系和密码子使用上比较接近.此外,来自日本的BVDV-1/No.12_E+和BVDV-1/No.12_E-,来自中国的BVDV-1/BJ1305和BVDV-1/BJ1308以及来自意大利的BVDV-1/VE/138cp/05和BVDV-1/VE/138ncp/05分别聚类于较小分支上,其结果和分子进化树结果一致.
3 讨论
瘟病毒基因组的遗传进化主要来自3个不同的过程:由病毒RNA依赖性RNA聚合酶的易错性导致的点突变的积累;非同源RNA的重组;同源RNA的重组[14].Weber MN等再对125个完整的瘟病毒序列分析后指出,其中2个BVDV-1、1个BVDV-2 和4个CSFV毒株的基因组是由同源重组进化而来[15].重组瘟病毒的存在对病毒分离株的系统发育分析和分型提出了挑战.虽然RNA重组是产生各种病毒变异体的主要动力,但越来越多的BVDV亚基因型的存在是随着时间积累的点突变的结果,也称为遗传漂移[16].因此,利用不同的方法对这些病毒进行分型研究,有助于揭示病毒进化的规律,具有重要意义.

图3 BVDV密码子COA数据聚类分析
目前,BVDV的分型研究受到了学者的广泛关注,主要是基于病毒基因组序列的分析结果对毒株进行分型,如5′UTR区域,以及Npro、E2等基因的序列特征.本研究以BVDV的RdRP基因和5′UTR序列分别进行了分子进化树的构建,基于这2种序列的BVDV分型结果基本一致,但在每个型内的不同分离株间的亲缘关系上存在较大差异.通常分子进化树的构建是基于瘟病毒属的RdRP基因序列进行的,二者产生差异的原因可能与5′UTR 序列在执行功能时的高级结构相关.此外,本研究利用基因密码子偏好性参数进行对应分析和聚类分析,结果显示,利用BVDV的RdRP序列以及5′UTR序列构建的分子进化树与BVDV密码子数据聚类分析结果相似,在型特异性上两种方法所得结果高度一致;COA数据分析显示,BVDV3个基因型的分布相对集中于不同象限中,其中BVDV-3基因型较为集中,BVDV-2和CSFV、BDV的COA数据分布更为接近,不同类型的BVDV在RSCU、密码子使用和氨基酸使用方面表现出了高度的型特异性.在每个BVDV型内部,不同分离毒株也具有相似的密码子偏好性,如分离于同一国家同种宿主动物的分离株具有很近的亲缘关系,密码子偏好性也相似.此外,来自亚洲不同国家的分离株,BVDV-1/AV69 VEDEVAC、BVDV-1/GX4、BVDV-1/KE9、BVDV-1/12F004和BVDV-1/BJ-2016也聚为一类,关系较近.值得注意的是,来自日本的BVDV-2/GBK_E-和来自美国的BVDV-2/USMARC-60767,来自意大利的BVDV-3/Italy-68/13cp和来自中国的BVDV-3/HN1507也分别聚在一起,反映了这几株病毒基因组在起源与演化过程中可能具有一定的关系.这将为研究基于密码子使用模式的系统进化分析提供更多的证据.除了上文提到的相同结果外,也有一些明显的例外.例如,BVDV-1/LETIZIA和BVDV-1/IZSUM在2种系统发育分析上结果一致,但在密码子聚类分析中二者各自聚为一类;还有BVDV-2/USMARC-60767和BVDV-2/USMARC-60779为同一地区同一宿主分离的病毒株,但在密码子使用偏性上却存在较大差异,其原因可能与BVDV-2/USMARC-60779基因组因测序不准确存在的15个不可翻译密码子有关.
生物体遗传信息的传递遵循“中心法则”,存在于DNA中的遗传信息在通过转录、翻译后,最终形成拥有生物功能的蛋白质[17].上述过程中最重要的一个环节是以三联体密码的形式编码蛋白质的结构单元-氨基酸[18].基因编码由64个密码子构成,但只有61个密码子编码20个标准氨基酸,其余3个为终止密码子,即TAA、TAG和TGA.由于遗传密码的简并性,大多数氨基酸由2-6个密码子编码,编码同一种氨基酸的密码子称为同义密码子.通常来说,某一物种或某一基因倾向于使用一种或几种特定的同义密码子.Ikemura曾指出高表达基因具有更强的密码子使用偏好性[19],还有一些研究揭示了密码子偏好性现象在进化速率估计和系统发育重建中的重要性[20].如前所述,部分病原微生物基因组密码子使用研究表明,密码子使用偏性与病原种类有密切关系,密码子使用对应分析数据的聚类分析,可以作为一种参考方法应用于物种鉴定中.
猜你喜欢
天津市教科院学报(2021年5期)2021-11-10
生物学通报(2021年9期)2021-07-01
生物学通报(2020年11期)2020-10-22
基层中医药(2020年5期)2020-09-11
发明与创新·中学生(2019年6期)2019-06-26
基层中医药(2018年5期)2018-08-31
中成药(2018年7期)2018-08-04
中国医学创新(2017年7期)2017-03-31
江苏农业科学(2016年8期)2017-02-15
制造技术与机床(2015年10期)2015-04-09
