
26例以皮疹为首发症状的儿童朗格汉斯细胞组织细胞增生症临床与病理分析
2020-07-01 08:23吴亚芬钱莹莹钱华苑春雨张婷李巍
中国中西医结合皮肤性病学杂志 2020年3期
吴亚芬,钱莹莹,钱华,苑春雨,张婷,李巍
(苏州大学附属儿童医院,江苏苏州215025)
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis,LCH)系指单核/巨噬细胞和树枝状细胞异常增生,并在不同组织中聚积的一组谱系疾病。LCH的临床分类方法多样,经典传统上分4型:勒-雪病(Letterer-Siwe disease,LSD),韩-雪-珂病(Hand-Schcüller-Chrislian disease,HSC),嗜酸性肉芽肿(Eosinophilic granuloma,EG)和先天性自愈性网状组织细胞增生症(Hashimoto-pritzker disease,HPD)[1]。LCH 好发于儿童,发病率约为 3%~5%[2]。LCH临床表现差异大,病情轻重不一,受累器官不同,预后各不相同。而以皮疹为首发症状的患儿,易发生漏诊、误诊。因此本文通过对26例组织病理及免疫组化结果确诊为LCH患儿的临床资料、组织病理、免疫组化结果进行系统分析,报道如下。
1 资料与方法
1.1 病例资料 收集2012年6月1日—2018年6月1日至苏州大学附属儿童医院皮肤科以皮疹为首发临床表现就诊,并经组织病理、免疫组织化学检查确诊为LCH的病例。26例病例中,男16例(61.54%),女 10例(38.46%),男女比例 1.6∶1;患儿年龄 40 d~7岁,平均年龄(20.4±4.7)个月,其中<1岁患儿16例(61.54%),1~2岁患儿 6例(23.08%),3岁1例,5岁2例,7岁1例;其中<1岁患儿中男女比例 2.2∶1,>1 岁患儿中男女比例 1∶1;病程为 10d~1 年。
1.2 方法 所有病例均行皮肤组织病理活检,常规石蜡处理、切片、HE 染色及 CD1a、S-l00、Ki-67 免疫组化染色。(一抗购自上海长岛公司,二抗及Ventana全自动免疫组化仪购自罗氏公司)所有抗体均设阳性对照,以磷酸盐缓冲液(PBS)代替一抗作为阴性对照。并调集患儿一般资料信息、临床照片、实验室检查、影像学检查、临床治疗信息进行系统回顾分析。若患儿某项数据缺失,进行相关统计分析时予以剔除。
2 结果
2.1 临床表现 皮损表现:26例患儿均以皮疹为首发症状就诊于我科,皮疹可发生于全身各部位,但最常见于躯干及头面部,其次为会阴部、臀部、肛周、口腔黏膜等处。皮疹形态各异,可呈脂溢性皮炎样表现,黄红或暗红色斑疹,上覆盖棕黄色鳞屑,见图1A。可呈紫癜样损害,变现为瘀点、瘀斑、出血性的丘疱疹,可伴有结痂,结痂后留下白斑,见图1B。可表现为结节性溃疡斑块,也可表现为泛发性的实质性、无痛性红棕色丘疹或结节,见图1C。少数患儿表现为局部肿物或外耳道脓性渗出。部分病例发热与皮疹之间有一定相关性,表现为发热时皮疹加重,并随发热皮疹反复。系统受累表现:24例患儿合并有其他系统受累,其中发热16例(61.54%),可呈持续发热或者间断发热,热型不定,骨损害10例(38.46%),其中最常见受累为颅骨6例,四肢长骨4例,主要表现为局部肿物,伴或不伴局部疼痛,一般无红肿及破溃。肝脾肿大15例(57.69%),肺部损害8例(30.77%),主要表现为咳嗽、咳痰、气促、呼吸困难等。其他症状还包括:突眼、贫血、尿崩症、齿龈肿胀坏死、牙齿松动或脱落,见表1。
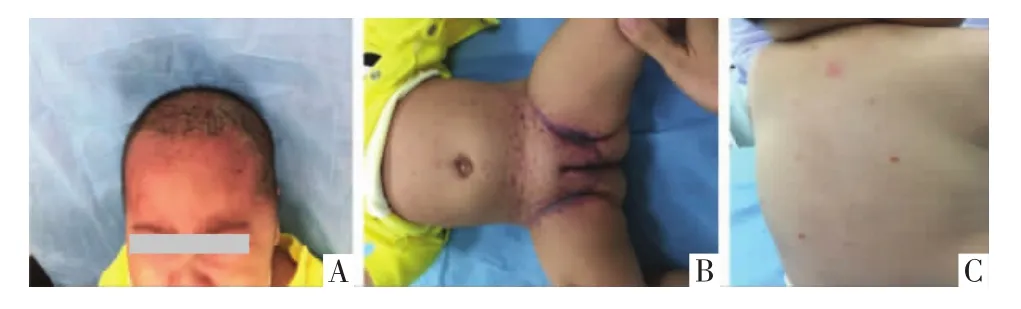
图1 朗格汉斯组织细胞组织细胞增生症患儿面部、躯干及四肢皮疹
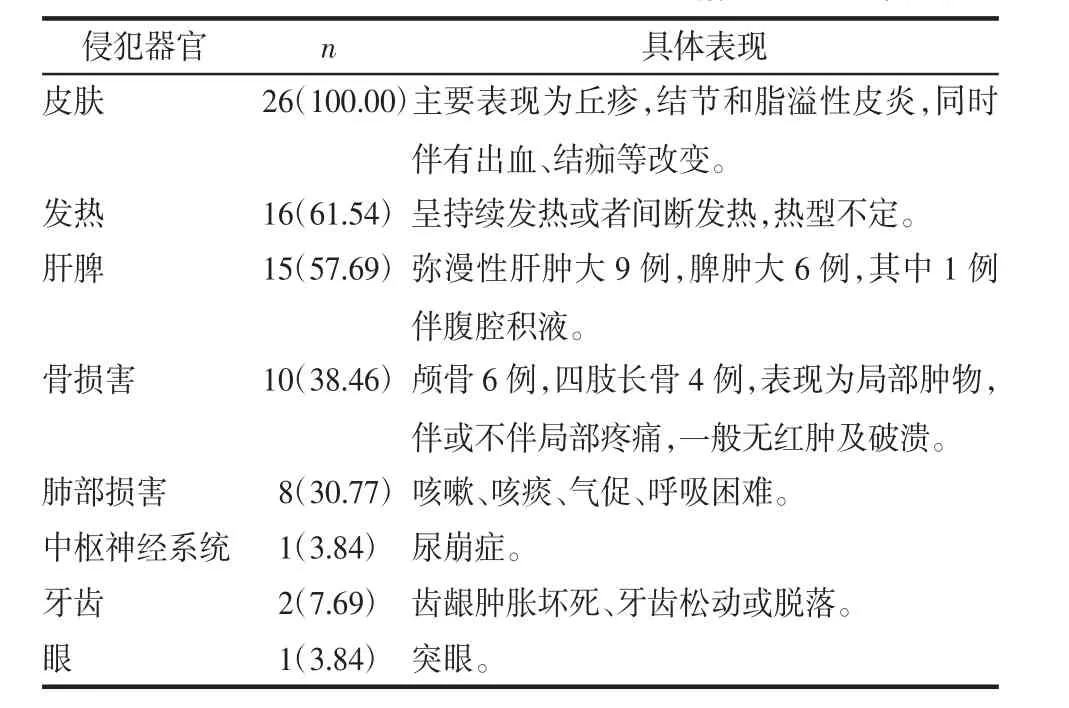
表1 26例患儿的临床表现情况 例(%)
2.2 实验室检查 血常规检查出现贫血8例,其中重度贫血2例,中度贫血3例,出现血小板减少5例,C反应蛋白增高10例,肝功能异常6例,丙氨酸转氨酶及天冬氨酸转氨酶升高5例,低蛋白血症4例,血清胆红素增高2例;8例患儿胸部X线:肺纹理增粗,呈网状或网点状阴影,晚期可表现为多发性囊肿及纤维化;骨骼X线检查或头颅CT检查:10例有溶骨性骨质破坏,颅骨受累6例,四肢长骨受累4例,见图2;骨髓涂片检查,表现为增生性骨髓象18例,见表2。
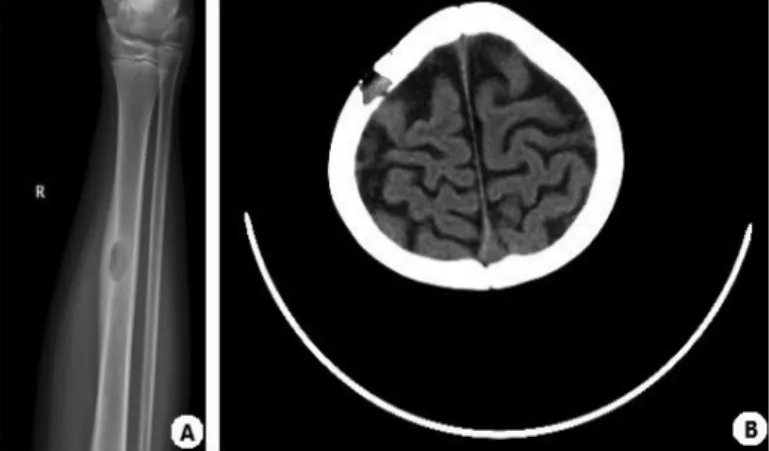
图2 朗格汉斯细胞组织细胞增生症患儿上肢X线及头颅CT影像图
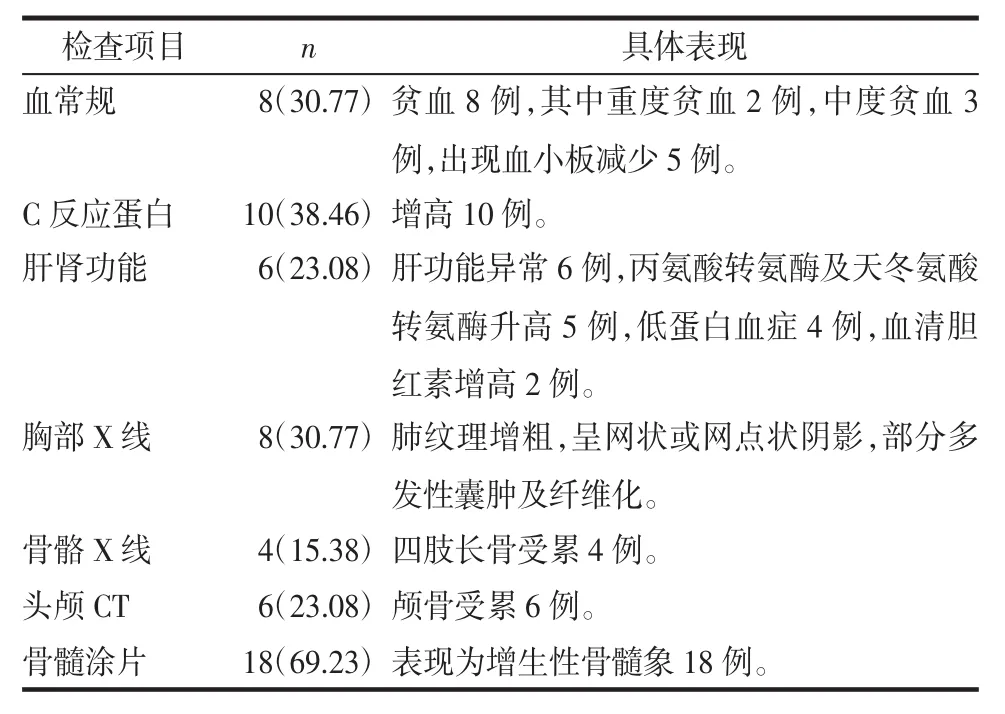
表2 26例患儿的实验室检查异常情况例(%)
2.3 组织病理学检查 26例均行组织病理学检查,可见真皮上部肿瘤细胞呈灶状或弥漫分布,典型的肿瘤细胞,细胞核染色浅,为肾形、分叶状或折叠呈锯齿形,胞质丰富,略呈嗜酸性。并有亲表皮性,有时可见Pautrier微脓肿样细胞聚集。真皮内还可见嗜酸性粒细胞、中性粒细胞和淋巴细胞浸润,见图3。26例均行免疫组化检查,CD1a(+)26例,S-100(+)26 例,Ki-67(平均 38.46%+),见图 4。
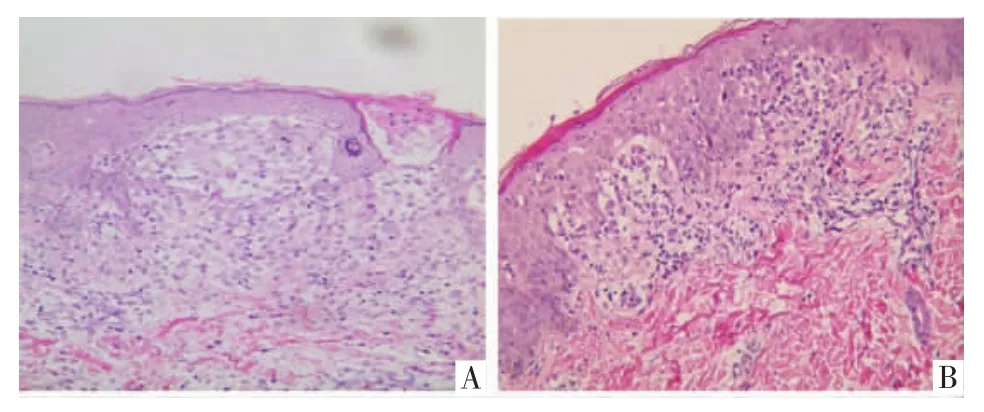
图3 朗格汉斯细胞组织细胞增生症患儿皮损组织病理像

图4 朗格汉斯细胞组织细胞增生症患儿皮损免疫组织化学像(×200)
2.4 治疗与预后 26例患儿均至我院血液科进行治疗,根据LCH累及系统及程度分类,单系统LCH(Single-system LCH,SS-LCH)和多系统 LCH(Multisystem LCH,MS-LCH),SS-LCH 有 1个脏器或系统受累(单病灶或多病灶),MS-LCH有≥2个脏器或系统受累,伴有或不伴有“危险器官”(肝脏、肺、脾脏或造血系统)受累[3]。其中2例为SS-LCH,24例为MS-LCH。根据Lavin-OsLand分级[4]制定化疗方案,主要治疗方案是泼尼松加长春新碱(Vincristine,VCR)化疗,18例化疗后好转,2例死亡,4例在化疗过程中未见明显改善,并发感染,2例放弃治疗。
3 讨论
LCH病因不明,有推测可能与代谢、病毒感染、免疫系统有关,2010年Allen等[5]为确定LCH病变中细胞特异性基因表达,采用全基因表达谱的对照研究,发现LCH细胞来源于骨髓树突状前体细胞,并能活化、募集T细胞至病灶部位,而非转化的表皮固有LC。近年来Badalian-Very等[6]在61例LCH患者中发现57%的患者BRAF V600E基因突变,Bubolz等[7]通过对653例样本进行Mata分析,显示在LCH中48.5%的患者BRAF V600E基因突变,BRAF是一种致癌基因,该发现为支持LCH为肿瘤性疾病提供有利证据。
LCH主要见于儿童,发病年龄高峰为1~3岁,男女比例为2∶1[8]。本研究中<3岁患儿共22例,男女比例为1.6∶1与文献报道相近,皮肤为LCH常见的受累部位,树叶等[9]对126例儿童LCH进行临床分析发现50%患儿有皮肤受累,而LCH的皮疹表现多样,极易与其他疾病相混淆。本文中26例患儿均以皮疹为首发临床表现就诊于我科,其中24例均以湿疹、脂溢性皮炎,传染性软疣、幼年黄色肉芽肿等治疗,首诊误诊率达92.31%。提示广大皮肤科医生,如临床中以湿疹、脂溢性皮炎治疗效果不佳的患儿应考虑LCH可能,需及时行皮肤活检。由于儿童皮肤活检难度较成人大,且家长难以接受有创性检查,儿童中很难进行常规病理活检,使得LCH早期皮肤诊断率较低。另外笔者发现急性起病的患儿,皮损多分布于头面部、躯干,主要变现为黄红或者暗红色斑丘疹,皮疹间常夹杂紫癜性损害,可出现渗出、出血、而后结痂脱屑,可留下来皮肤萎缩及色素脱失斑。慢性发病的患儿皮损可分布于身体各处,常表现为圆形或者椭圆形,黄红色、黄色丘疹或结节。
研究中发现,<1岁的患儿多为LSD,一般受累器官>2个,且病情重,进展快,随着年龄增长,受累器官减少,病情较轻。本组病例中骨受累10例(38.46%),其中颅骨6例,长骨4例,头颅CT表现为圆形或者椭圆形虫蚀缺损,而长骨X线可见边缘不规则的溶骨性损害,大龄儿童多见,常表现为痛性肿胀或无症状。本研究中肝脾累及率达57.7%,常见于多器官累及,且病情进展较快的患儿,可出现肝功能异常、黄疸、门脉高压、肝衰竭等表现,因此在皮肤科就诊患儿中,如皮疹考虑LCH,可先行无创性的肝脾B超检查,如发现异常,应高度怀疑是否为LCH,在本研究的26例患儿中,8例患儿有肺部累及,临床上主要表现为咳嗽、咳痰、气促、呼吸困难等,胸部X线主要表现为肺纹理增多增粗,弥漫性斑状、点状、栗粒状阴影。其中值得注意的是由于骨损害累及耳乳突,有2例患儿表现为顽固性中耳炎,反复外耳道脓性分泌物,抗生素治疗效果不佳,另外内分泌系统受累最常见的是尿崩症,本研究中仅有1例患儿。
LCH诊断标准[10]:初步诊断:组织病理学的光镜下可见到肿瘤浸润;明确诊断:光镜检查所见加以下4项指标2项或2项以上阳性:ATP酶、S-100、α-D-甘露糖酶、花生凝集素。最佳诊断:光镜所见加电镜下发现病变细胞内有Birbeck颗粒和(或)CD1a单抗染色阳性。CD1a是诊断LCH常用的标志物,敏感性和特异性均高,定位于细胞膜,本组26例CD1a染均为阳性。S-100敏感度高,但特性差,一般需联合CD1a或者Langerin可明确诊断。本组26例S-100均为阳性,此外皮疹相关特异性检查还包括皮疹印片和皮疹电镜检查。
LCH是一组异质性疾病,根据受累器官及Lavin-Osband分级,制定个体化治疗方案极为重要,近年来儿童的LCH的治疗和预后得到很大程度的改善,本文中26例患儿均以皮疹为首发症状就诊于皮肤科,而首诊误诊率达92.31%,至我科就诊时,因反复治疗效果不佳,均行皮肤活检,进而确诊,因此提高皮肤活检率仍是降低LCH误诊率的重要手段,同时应加强完善各项实验室检查,以明确诊断尽早治疗。
猜你喜欢
读者·校园版(2020年14期)2020-07-16
优雅(2019年10期)2019-11-17
环球市场信息导报(2018年1期)2018-05-30
爆笑show(2015年5期)2015-07-09
环球时报(2015-03-21)2015-03-21
中国民族民间医药·下半月(2011年4期)2011-09-27
文化月刊·遗产(2009年3期)2009-08-11
幸福·悦读(2009年2期)2009-03-17
中学英语之友·高一版(2008年3期)2008-04-10
浙江中医杂志(2004年10期)2004-03-08
