
MOLECULAR CHARACTERIZATION OF TWO VARIANT TOTIVIRUSES ISOLATED FROM CULEX TRITAENIORHYNCHUS IN CHINA
2020-06-05 11:06PENGHongHongWANGYuanYuanDAIKeZHAOQiuMinZUOShuQingTONGYiGangZHANGJiuSong
寄生虫与医学昆虫学报 2020年1期
PENG Hong-Hong WANG Yuan-Yuan DAI Ke ZHAO Qiu-Min ZUO Shu-Qing TONG Yi-Gang,3 ZHANG Jiu-Song*
(1. Anhui Medical University, Hefei 230000, China; 2. State Key Laboratory of Pathogen and Biosecurity, Institute of Microbiology and Epidemiology, AMMS, Beijing 100071, China; 3. Beijing Advanced Innovation Center for Soft Matter Science and Engineering (BAIC-SM), College of Life Science and Technology, Beijing University of Chemical Technology, Beijing 100029, China)
Abstract Two totiviruses, temporarily named Totivirus ND3 (TOV_ND3) and FD17(TOV_FD17), were isolated from Culex tritaeniorhynchus captured in Shanghai City. The full sequences were determined using high-throughput sequencing method. The results showed that the genomes of TOV_ND3 and FD17 were 7 612 and 7 611 nucleotides, respectively in length, and encoded a capsid protein and an RNA-dependent RNA polymerase. The nucleotide sequence of TOV_ND3 had the highest similarity to Cx. tritaeniorhynchus totivirus NJ2, while the FD17 was most similar to Omono River virus strain AK4, with 84.6% and 84.9% identity, respectively, and 74.1% identity between themselves. Phylogenetic analysis revealed that the two newly obtained viruses were grouped into the unassigned totiviruses. Our results suggested that the two new viruses were variant totiviruses.
Key words Totivirus; Virus isolation; High-throughput sequencing; Complete genome
Members of family Totiviridae belonged to nonsegmented dsRNA virus and included five assigned virus genera,Totivirus,Victorivirus,Trichomonasvirus,GiardiavirusandLeishmaniavirus, containing 28 virus species, such asSaccharomycescerevisiaevirus,Helminthosporiumvictoriaevirus,Trichomonasvaginalisvirus,Giardialambliavirus, Leishmania RNA virus (Ghabrialetal., 2009; Goodmanetal., 2011; Dunnetal., 2013; Janssenetal., 2015; Rodriguez-Cousinoetal., 2017). The genome of the totivirus was composed of 4.6-7.6 kb in length and encoded a capsid protein (CP) and an RNA-dependent RNA polymerase (RdRp) (Ghabrialetal., 2009; de Limaetal., 2019). The viruses were generally reported to infect fungi, parasites and protozoa (Ghabrialetal., 2009; Goodmanetal., 2011; Dunnetal., 2013; Janssenetal., 2015). In addition, some unassigned totiviruses were generally isolated from plant, fish, shrimp and arthropods, such as Maize associated totivirus from plant, Golden shiner totivirus from fish, Penaeid shrimp infectious myonecrosis virus from shrimp, Lonestar tick totivirus from ticks(Poulosetal., 2006; Moretal., 2016).
So far, only five totiviruses were isolated from mosquitoes, including Omono River virus (ORV) AK4 and Y61 in Japan,Armigeressubalbatusvirus SaX06-AK20 (AsTV),Culextritaeniorhynchustotivirus NJ2 (CTV_NJ2) and NJ3 (CTV_NJ3) in China. In 2006, AsTV was first isolated fromAr.subalbatusin Shaanxi Province(Zhaietal., 2010), and identified as a new species in Totiviridae. In 2011, ORV-AK4 and ORV-Y61 were isolated fromCulexmosquitoes in Japan (Isawaetal., 2011). Then in 2012, ToV-TJ was first isolated from bat feces in Tianjin city and Hainan Province(Yangetal., 2012). At the same year, ToV-TJ was also found fromCx.tritaeniorhynchuscollected in Yunnan province(Guoetal., 2012). During a survey of Japanese encephalitis virus (JEV) in Shanghai City, eastern China, in 2007, two totiviruses were found fromCx.tritaeniorhynchus. Their molecular characteristics were reported in this study.
1 Materials and methods
A total of 5 220 mosquitoes (allCx.tritaeniorhynchus) were collected during the mosquito activity season in 2007, using ultraviolet light lamps (Wuhan Lucky Star Environmental Protection Technology Co., China) placed beside pigs and livestock houses in the villages of FX and NH district, Shanghai city. Collected mosquitoes were grouped into 102 separate pools with approximately 50-100 mosquitoes based on collection sites, and stored in liquid nitrogen until test. Each mosquito pool was homogenized with RPMI-1640 medium containing 2% fetal calf serum, and then centrifuged. The supernatants were inoculated onAedesalbopictusC6/36 cell lines for viral isolation. Cytopathic effect (CPE) was observed daily for 7 days. The culture supernatant was collected as a positive viral isolate if it showed CPE in three successive cell passages (Zuoetal., 2014).

Fig.1 Cytopathic effect on C6/36 cells caused by TOV_FD17A. Infected C6/36 cells; B. Uninfected C6/36 cells.
Total RNA was extracted from the positive isolate using a High Pure Viral RNA Kit (Roche, Switzerland) and used to construct a cDNA library using Ion Total RNA-Seq Kit v2 (Thermo Fisher Scientific,USA) according to the manufacturers’ instructions. The library was then subjected to high-throughput sequencing. To remove low-quality reads, the FASTP was used to filter the raw data. Virus-related sequences were discovered and compared to GenBank nucleotide database using BLAST program. CTV_NJ2 (GenBank accession no. KX456218.1) and ORV-AK4 (GenBank accession no. AB555545) were used as references for construction of viral reads. Cytoscape v2.8.3 software and the ContigScape plugin were used to identify relationships among contigs(Smootetal., 2010; Tangetal., 2013). The complete genomes were annotated by Rapid Annotation using Subsystem Technology (RAST, http://rast.nmpdr.org) (Azizetal., 2008). Nucleotide sequences were compared by BLAST method. Multiple sequence alignments and phylogenetic analysis were carried out using the MEGA 7.0 program (Kumaretal., 2016) based on the two newly obtained sequences and 19 reference sequences, including the members of the five genera and 14 representative unclassified totiviruses (Tab. 1). Phylogenetic tree was constructed from aligned nucleotide sequences by using the Maximum-Likelihood method with bootstraps values calculated from 1 000 replicates.
2 Results
Of five positive isolates, three were identified as JEV by reverse transcription-polymerase chain reaction (RT-PCR) with the virus-specific primers, but no any positive results were obtained in the other two isolates by traditional PCR methods with genus-specific primers for arboviruses in 2007. In order to identify the isolates, a retrospective study was conducted by high-throughput sequencing method in 2018.

Tab.1 Information of the totivirus strains used in the phylogenetic analysis in the study
The CPE caused by the two positive isolates were similar on C6/36 cells. It could be rapidly observed within 24 hours after inoculation, and was characterized with cell fusion and disintegration (Fig. 1).
The results of high-throughput sequencing indicated that the two isolates were related to totiviruses, temporarily named as TOV_ND3 and TOV_FD17 according to collection sites of mosquito samples. The full genomic sequence of TOV_ND3 was 7 612 nucleotides in length and contained two open reading frames (ORFs) encoded a putative CP with 1 685 aa located from nucleotide (nt) position (7-5 064 nt) and an RdRp with 738 aa (5 319-7 535 nt). The TOV_FD17 genome was consisted of 7 611 nt and two ORFs encoded a 1 685 aa of CP (7-5 064 nt) and a 764 aa of RdRp (5 226-7 535 nt). The G+C content of the genomic sequences of TOV_ND3 and TOV_FD17 were 51% and 49%, respectively. The newly obtained sequences had been deposited in GenBank with the accession number MN196674 and MN196675. The nucleotide identity between two new sequences was 74.1%. TOV_ND3 and TOV_FD17 had the highest identity to CTV_NJ2 (84.6%) and ORV-AK4 (84.9%), respectively, and then with TOV-TJ, ORV-Y61, ORV-TB94, CTV_NJ3 andDrosophilamelanogastertotivirus (DTV) (65.6%-84.7%). Comparison results of nucleotide sequences among the new isolates and these viruses were shown in
Tab. 2. The identities of amino acid sequences of CP and RdRp were 77.2%-95.9% and 81.1%-98.9% among the two new viruses and above seven viruses, respectively. The similarity of CP and RdRp between themselves were 84.3% and 87.1%. The new viruses had low similarity of nucleotide and amino acid sequences when compared with the classified totiviruses.
To illustrate the genetic relationships among the two variant totiviruses and other reference members of the family Totiviridae, a phylogenetic tree was constructed based on the complete nucleotide sequences (Fig. 2). All viruses used in the tree were grouped in three clusters. Five assigned totiviruses represented different virus genera were located in separate clade of the two clusters, whereas 13 were grouped into another large cluster, including TOV_ND3 and TOV_FD17. The TOV_ND3 was most closely related to CTV_NJ2, while TOV_FD17 was closed to ORV-AK4 and TOV-TJ, which were consistent with the comparison results of nucleotide sequence identity. The TOV_ND3 and TOV_FD17, together with TOV-TJ, CTV_NJ2, ORV-AK4, ORV-Y61, ORV-TB94 and ORV-TB102 were closed in terms of their phylogenetic placement.
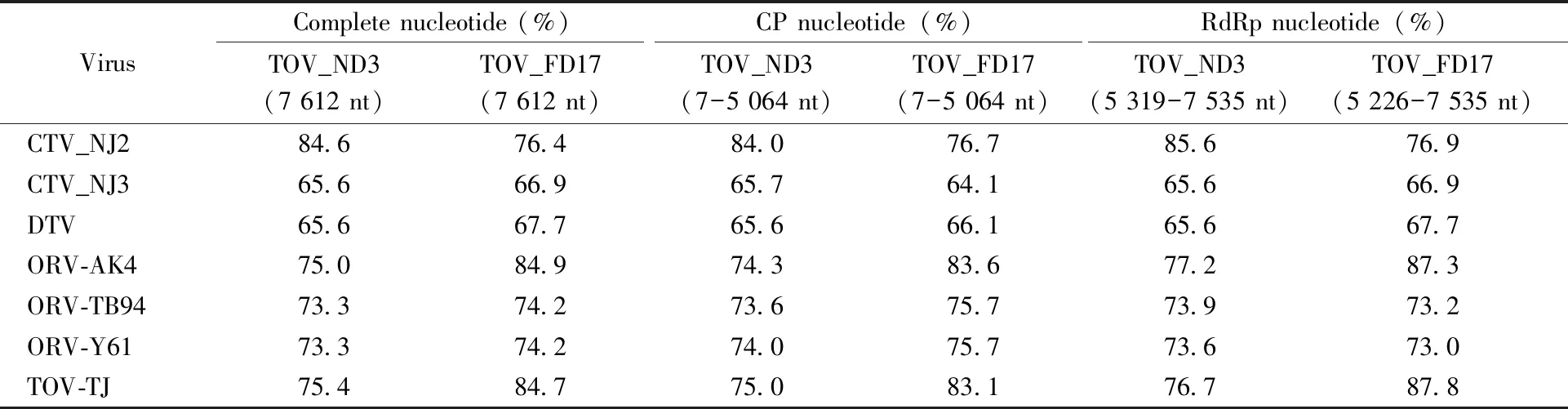
Tab.2 Comparison results of nucleotide sequences among the new isolates and several totiviruses
CTV_NJ2:Culextritaeniorhynchustotivirus NJ2; CTV_NJ3:Culextritaeniorhynchustotivirus NJ3; DTV:Drosophilamelanogastertotivirus; ORV-AK4: Omono River virus AK4; ORV-TB94: Omono River virus TB94; ORV-Y61: Omono River virus Y61; TOV-TJ: Tianjin totivirus.
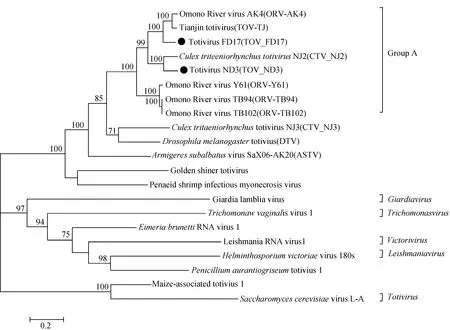
Fig.2 Phylogenetic analysis based on 21 complete nucleotide sequences of totiviruses The phylogenetic tree were constructed by using the Maximum-Likelihood method based on the JTT matrix-based model. The TOV_ND3 and TOV_FD17 are marked with solid black rings.
3 Discussion
In the present study, two totiviruses, TOV_ND3 and TOV_FD17, were identified fromCulextritaeniorhynchuscaptured in Shanghai City, eastern China. The CPE caused by the viruses on C6/36 cells were characterized with cell fusion and disintegration, which were consistent with those caused by Tianjin totivirus (TOV-TJ) isolated from bat faces in China(Yangetal., 2012). The TOV_ND3 had the highest similarity of nucleotide sequence to CTV_NJ2(only 85%)isolated fromCx.tritaeniorhynchusin Yunnan province, southwestern China (Huangetal., 2018), and TOV_FD17 was the most close to ORV-AK4 and TOV-TJ isolated fromCulexmosquitoes in Japan and China (Isawaetal., 2011; Yangetal., 2012), with approximate 85% identity, indicating that TOV_ND3 and TOV_FD17 were variant totiviruses. Additionally, the genetic similarity among these viruses and other related totiviruses covered from 66% to 85%, suggesting genetic diversity of totiviruses. Phylogenetic analysis showed that these viruses, together with CTV_NJ2, ORV-AK4, ORV-Y61, ORV-TB94 and ORV-TB102, were grouped into one cluster named as unclassified totiviruses (Fig.2, Group A). All of them were reported from arthropods especiallyCulexmosquitoes(Isawaetal., 2011; Huangetal., 2018), suggesting thatCulexmosquitoes may be the main vector of the unclassified totiviruses. These totiviruses should be proposed as a new genus and assigned to family Totiviridae, as suggested by Zhaietal. (Zhaietal., 2010). So far, totiviruses from mosquitoes were reported in many regions, including Shaanxi province, Yunnan province, Hainan province and Tianjin city, which indicated the wide distribution of totiviruses carried by mosquitoes in China.
In conclusions, two variant totiviruses were obtained from mosquitoes, in which few totiviruses were reported. Our findings enriched the information of family Totiviridae.
NucleotidesequenceaccessionnumberThe complete genome sequences of TOV_ND3 and TOV_FD17 with annotation were deposited in the NCBI nucleotide database under the accession number MN196674 and MN196675.
FundingThis research was supported by grants from the National Natural Science Foundation of China (No. 81273138 and No. 81572045).
ConflictofinterestThe authors declare that they have no conflict of interest.
EthicalapprovalThis article does not contain any studies with human participants or animals performed by any of the authors.
