
一个发作性运动诱发性运动障碍家系的遗传学诊断
2020-04-15 08:43:26胡新超毛澄源史长河范丽媛胡正威杨志华许予明
郑州大学学报(医学版) 2020年2期
胡新超,毛澄源,史长河,范丽媛,张 槊,胡正威,杨志华,范 雨,杨 靖,许予明
郑州大学第一附属医院神经内科 郑州 450052
发作性运动诱发性运动障碍(paroxysmal kinesigenic dyskinesia,PKD)是阵发性运动障碍疾病最常见的一种类型。PKD的临床特点为发作性运动诱发的反复不自主动作,每次持续时间不超过1 min,对小剂量卡马西平或抗癫痫药治疗有效[1]。PKD发病率很低(约为1∶150 000),具有自愈倾向,患者常于儿童或青少年期发病,成年后症状缓解至完全正常[2]。大多数PKD呈常染色体显性遗传,存在不完全外显性,少部分PKD散发存在。最初,位于16号染色体的EKD1区(16p11.2-q12.1)[3]、EKD2区 (16q13-q22.1)[4]和非16号染色体的EKD3区[5]被认为与家族性PKD有关。随后,3项独立的研究[6-8]锁定位于16p11.2的PRRT2基因为家族性PKD的致病基因。目前,国内已鉴定出48个PKD家系,其中绝大多数家系携带PRRT2 c.649dupC(p.R217PfsX8),此外还存在c.487C>T(p.Q163X)、c.649_650InsC(p.P217fsX7)和c.796C>T(p.R266W)等20多种致病突变[9]。本研究纳入了一个PKD家系,总结了该家系的临床特征并进行基因检测以协助临床诊断和治疗。
1 对象与方法
1.1研究对象先证者(Ⅲ-2),男,20岁。以“发作性四肢僵硬扭曲12 a”为主诉至我院就诊。患者7岁时常于精神紧张状态下突然运动时出现双手屈曲成固定形状,意识清楚伴兴奋感,严重时伴面部不可抑大表情;每次持续数秒后自行缓解,每日发作数次;情绪紧张时发作次数明显增多,心情放松时减少。对小剂量卡马西平治疗有效。无癫痫、偏头痛、婴幼儿惊厥等神经系统疾病史。先证者既往被诊断为“精神心理疾病”“幼儿多动症”。神经系统查体:神志清楚,对答流利,未发现阳性体征。辅助检查:EEG和颅脑MRI未见异常。
收集2019年1月至10月至我院体检科检查的健康人作为正常对照。纳入标准:年龄5~50岁;既往体健;签署知情同意书。排除标准:有婴儿惊厥、癫痫、偏头痛及其他神经系统疾病史。
1.2家系调查该家系(汉族)三代18人共有5例患者,见图1。先证者母亲(Ⅱ-2),女,48岁。15岁常于他人呼叫提问时出现无法控制的四肢不自主扭曲,每次持续不超过10 s后自行缓解,至24岁时症状自行好转。患者症状不影响日常生活,未曾接受系统治疗。无偏头痛、癫痫及其他神经系统疾病史。神经系统查体未发现阳性体征。辅助检查:EEG和颅脑MRI未见异常。
先证者姨妈(Ⅱ-8),女,42岁。14岁时出现与先证者及其母亲相同症状,25岁症状完全消失,未曾接受系统治疗。先证者2个双胞胎表弟(Ⅲ-7和Ⅲ-8),男,12岁,均于6岁时出现与先证者相同症状。
此外,先证者姥爷(Ⅰ-1),男,80岁,既往有被害妄想病史,已治愈。先证者姥姥已故(Ⅰ-2),既往于情绪激动时出现四肢抽搐,意识清楚,不伴口吐白沫,给予对症治疗后缓解(具体不详)。
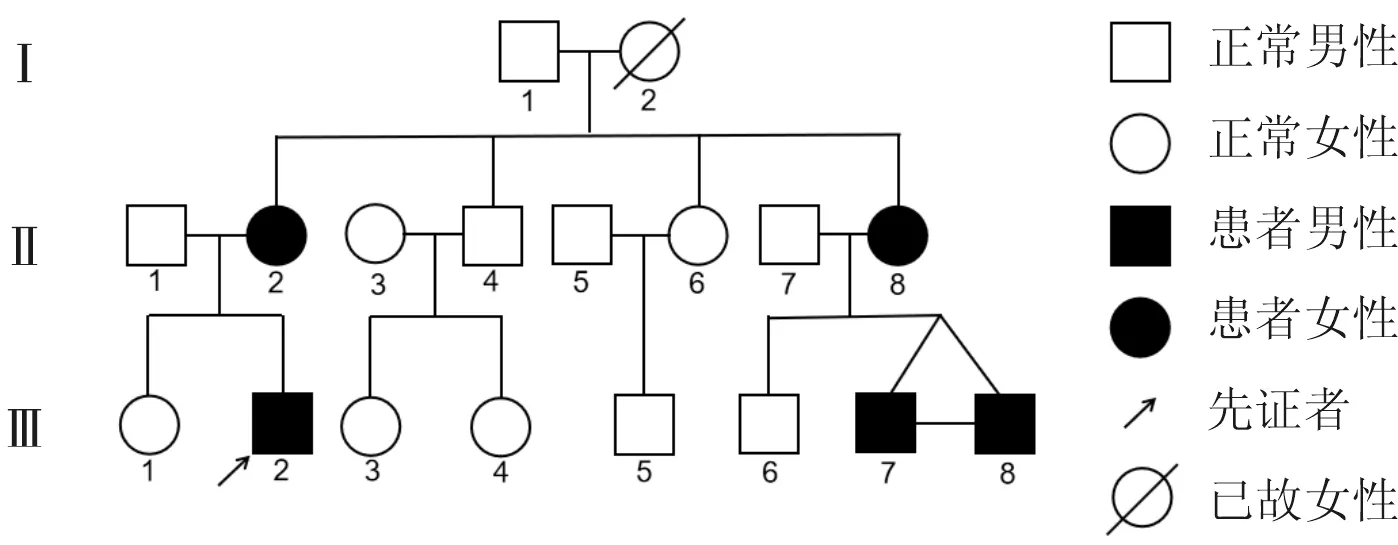
图1 PKD家族谱系图
1.3基因组DNA的提取所有患者及先证者父亲签署知情同意书后,采集外周血5 mL于EDTA抗凝管。同时抽取200名正常对照的外周血。采用全血基因组DNA 提取试剂盒(北京康为世纪生物科技有限公司)提取基因组DNA。Thermo ScientificTMNanoDropTMOne超微量紫外分光光度计检测DNA纯度和浓度。提取的DNA样本于-20 ℃保存。
1.4目标序列捕获测序先证者DNA样本送至北京金准基因科技有限责任公司进行基因包目标序列捕获测序。全自动超声破碎仪(新芝SCIENTZ08-Ⅲ)超声打断DNA样本,得到200~300 bp的DNA片段。采用KAPA Library Preparation Kit构建DNA文库。对目标基因编码区及临近剪切区的DNA进行捕获和富集。捕获后的样本进行Illumina NovaSeq高通量测序。
1.5Sanger测序验证对家系内所有患者、先证者父亲及200名正常对照的目标点进行验证。采用Primer Premier 6.0软件设计PCR引物,上游引物序列5’-AGCCCCACCTGAACCAG-3’,下游引物序列5’-CAGAGGCTCTATTGCAGCG-3’。PCR反应体系(30 μL):2×PCR Master Mix 12 μL,上下游引物各1 μL,DNA 模板 1 μL,ddH2O 15 μL。正向测序PCR条件:95 ℃预变性5 min;95 ℃变性30 s,65 ℃退火30 s,72 ℃延伸30 s,25个循环,每循环降低0.6 ℃。反向测序PCR条件:95 ℃变性30 s,50 ℃退火30 s,72 ℃延伸1 min,20个循环;72 ℃延伸10 min。PCR产物经15 g/L琼脂糖凝胶电泳鉴定片段大小后进行Sanger测序,测序结果与家系内正常人(先证者父亲)和200名正常对照进行比对。
2 结果
2.1家系临床特征本家系所有患者症状相似,具有以下特点:①突然运动改变、压力等诱发肢体屈曲动作。②发作时意识清楚。③每次持续时间不超过1 min,每日发作数次至十数次。④发病年龄1~20岁。⑤成年后症状完全缓解。⑥对小剂量卡马西平和抗癫痫药物治疗有效。⑦排除神经系统其他疾病。该家系符合单纯型PKD的诊断标准。与Ⅱ代患者相比,Ⅲ代患者发病年龄更早,症状更明显。
2.2遗传学分析目标序列捕获测序发现先证者PRRT2外显子2存在一处杂合突变:c.535 C>T,导致 p.Q179* 氨基酸改变(谷氨酰胺>终止)。查询HGMDpro 数据库未发现该突变的报道。美国医学遗传学和基因组学院对PRRT2 c.535 C>T评级为疑似致病突变。Sanger测序显示家系内其余患者均携带此突变,家系内正常人(先证者父亲)未发现该位点突变,符合家系内共分离,见图2。此外,200名正常对照未发现PRRT2 c.535C>T的存在,证实了PRRT2 c.535 C>T(p.Q179*)是该单纯型PKD家系的致病突变。

上:先证者PRRT2 c.535C>T;中:先证者母亲PRRT2 c.535 C>T;下:先证者父亲正常
图2Sanger测序结果
3 讨论
PRRT2基因被报道与家族性PKD、家族性婴儿惊厥伴发作性舞蹈手足徐动症、良性家族性新生儿癫痫2型有关[10]。目前国内外已发现的PRRT2突变大约有81种,其中包括36个截短突变、29个错义突变、3个延伸突变、6个剪接位点改变以及7个导致PRRT2完全缺失的改变[11]。这些突变大多位于第2和第3外显子。此外,内含子的剪切位点致病突变也有报道[12]。
PRRT2基因有4个外显子,编码含340个氨基酸的富含脯氨酸的跨膜蛋白(PRRT2蛋白)。PRRT2蛋白含有两个胞外结构域、两个跨膜结构域和一个胞内结构域[11],主要定位于谷氨酸能轴突的突触前膜,通过以下途径负调节突触传递:①与SNARE复合体联结并参与SNARE复合体的组装[13-14]。②与SNAP25蛋白和突触标签蛋白1/2相互作用,共同调节Ca2+依赖的突触囊泡胞吐[15-16]。③调节Nav1.2/Nav1.6通道介导的神经元兴奋[17]。无义突变使PRRT2蛋白的翻译提前终止,截断氨基末端的胞外区肽链并丧失羧基末端的跨膜结构域[8]。截短的PRRT2蛋白不能锚定在细胞膜上而丧失正常的生理功能,引起神经元兴奋性紊乱,从而触发PKD样症状。另一方面,截短的PRRT2 mRNA可被细胞内保护途径无义突变介导的mRNA降解途径降解,导致PRRT2单倍体不足,同样可影响蛋白质的功能而致病[18-19]。此外,PRRT2的无义突变被报道[20]与神经元迁移能力减弱和神经发育迟滞有关。小鼠脑组织PRRT2 mRNA水平在早期发育阶段逐渐升高而成年后下降[8],人脑中有类似结果[11]。这与PKD患者青少年时期发病并且成年后有自愈倾向的特点一致。目前,PKD的发病机制尚未完全明晰,其机制仍值得进一步探究。
本研究中,我们在一个中国汉族单纯型PKD家系中发现了一个未曾报道的PRRT2 c.535 C>T(p.Q179*)致病突变,这一发现拓宽了PRRT2相关PKD的突变谱范围。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22 02:52:50
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
广州大学学报(自然科学版)(2019年1期)2019-05-07 01:33:26
天津科技大学学报(2016年1期)2016-02-28 16:59:45
广东海洋大学学报(2015年4期)2016-01-13 08:39:30
湖北师范大学学报(自然科学版)(2015年2期)2016-01-10 08:41:53
听力学及言语疾病杂志(2015年5期)2015-12-24 01:47:04
首都医科大学学报(2015年4期)2015-12-16 13:00:08
重庆医学(2015年12期)2015-03-05 05:52:54
