
TSR1突变导致先天性白内障及其在晶状体中的表达
2020-04-13 06:30:56于雅洁邱峰张新安
遗传 2020年2期
于雅洁,邱峰,张新安
研究报告
突变导致先天性白内障及其在晶状体中的表达
于雅洁1,2,邱峰3,张新安1
1. 沈阳体育学院运动人体科学学院,沈阳 110102 2. 辽宁省金秋医院,沈阳 110016 3. 沈阳市第四人民医院,沈阳市眼科医院,沈阳 110031
先天性白内障(congenital cataract, CC)是一种罕见的晶状体发育异常疾病,主要表现为晶状体部分或完全浑浊。先天性白内障遗传异质性高,已鉴定的致病基因多达266个。本研究在一个中国先天性白内障家系中通过全基因组测序及Sanger测序验证,筛查到一个新的先天性白内障候选致病基因,与家系疾病表型共分离。通过minigene实验证实该变异影响基因mRNA剪接。Western blotting、免疫荧光和RT-PCR实验证实在人晶状体上皮细胞SRA01/04、年龄相关性白内障患者晶状体前囊膜组织、24周人胎眼晶状体和小鼠晶状体中表达。通过对iSyTE数据库的分析发现,在小鼠的胚胎期和不同发育时期的晶状体中都有表达,且在晶状体特异性CBP:p300双敲除小鼠中表达下调。提取在CBP:p300双敲除小鼠晶状体中与具有相同表达模式的一组基因进行蛋白质-蛋白质相互作用网络(protein-protein interaction,PPI)分析,结果表明筛选出6个基因与存在直接相互作用。GO功能分析表明Tsr1参与核糖体的组装,还可能在MAPK-Erk信号通路中发挥作用,为进一步明确在晶状体中的功能提供了有价值的研究线索。
TSR1;晶状体;蛋白质相互作用网络
先天性白内障(congenital cataract,CC)是指出生前即存在或出生后1年内才逐渐形成的白内障,发病率为0.1‰~0.6‰,是一种较为常见的儿童眼病,是小儿失明和弱视的主要原因[1,2]。先天性白内障通常表现为晶状体全部或部分浑浊,常伴随一种或多种其他的眼部并发症,如斜视、眼球震颤、先天性小眼球、眼内组织异常和缺失、视网膜和脉络膜病变等。先天性白内障发病受遗传和环境因素影响,遗传性先天性白内障多为单基因突变导致并且具有很强的遗传异质性。遗传方式包括常染色体显性遗传、常染色体隐性遗传、X连锁遗传,其中以常染色体显性遗传居多。目前已经报道的先天性白内障致病基因多达266个,其中32个基因可以导致单纯性先天性白内障,多数基因导致综合征性先天性白内障[3~6],除白内障表型外还包括一些眼部其他表型和身体其他组织和器官的异常和病变,如Warburg- Micro综合征、Zellweger综合征、Wolfram-like 综合征和Nance-Horan综合征等[7~10]。
本研究利用全基因组测序技术在一个中国先天性白内障家系中筛查致病基因突变,并利用minigene、RT-PCR、免疫荧光、Western blotting和生物信息学分析探讨致病可能机制。
1 材料与方法
1.1 材料
本研究的家系样本为沈阳市第四人民医院眼科收集,3代共8名患者,遗传方式为常染色体显性遗传,所有参与者均接受了裂隙灯检查。正常人群为炎黄中国人基因频率数据库(Chinese millionome database, CMDB)和北京诺禾致源科技股份有限公司(NovoDb)的中国正常人群样本库及中国医科大学附属第四医院体检中心的105例正常人,年龄相关性白内障患者晶状体前囊膜组织和24周人胎眼晶状体组织的石蜡切片由中国医科大学附属第四医院眼科中心提供,人晶状体上皮细胞SRA01/04为实验室自存,使用的小鼠为野生型C57BL/6,生物信息学数据分析来自于CBP:p300双敲除小鼠模型。本研究通过了沈阳市第四人民医院和中国医科大学的伦理审查,所有参与者均签署知情同意书。
1.2 基因组DNA提取及全外显子组测序
使用TIANamp Genomic DNA Kit(北京天根生化科技有限公司)提取先天性白内障家系成员基因组DNA,选择2名患者(III-3和III-5)和1名正常人(II-2)进行全外显子组测序。
1.3 全外显子组测序结果分析
全外显子组测序原始数据由BWA软件mem命令比对到hg19参考基因组上,比对结果由samtools软件进行排序、标记重复和格式转换生成bam格式文件并构建索引,使用GATK4.0软件进行snp-calling生成vcf格式储存的变异信息文件,使用annovar软件对变异进行注释,首先将所有变异与ExAC、dbSNP、EVS、GnomAD、1000genomes、CMDB和NovoDb等数据库进行比对,排除正常人群中出现频率高于0.01%的变异,使用基因组可视化软件Integrative Genomics Viewer (IGV)对位于外显子、外显子非翻译区(untranslated region, UTR)和剪接位点的变异进行可视化,排除假阳性结果,剩余位点作为候选变异。
1.4 PCR-Sanger测序及突变预测
对全外显子组测序数据进行分析筛选,设计特异性引物扩增所有候选位点并进行Sanger测序验证。使用MEGA-X软件进行位点保守性分析,Human splicing finder 3.1在线软件预测突变对剪接的影响。
1.5 minigene实验
设计引物扩增第1内含子3¢端150 bp序列以及第2外显子、第2内含子、第3外显子、第3内含子、第4外显子和第4内含子5¢端150 bp序列,上游引物TSR1-BamF:5¢-CGCGGATCCGCTATTTGGCCGTTATTAACTCTT-3¢,下游引物TSR1- MluR:5¢-CGACGCGTAAGCCCAACACTGAACTACCC-3¢,以先证者基因组DNA为模板,PCR反应条件为98℃预变性2 min,98℃变性20 s、60℃退火20 s、72℃延伸90 s,35个循环,72℃延伸5 min。经HⅠ和Ⅰ酶切后连入pCAS2质粒,构建野生型和突变型minigene质粒。pCAS2空载、TSR1野生型和TSR1突变型质粒转染至SRA01/04细 胞,24 h后收集细胞使用Trizol(北京全式金生物 技术有限公司)提取总RNA并反转录成cDNA(北京全式金生物技术有限公司),使用载体特异性引物(pCAS2-RTF:5¢-CTGACCCTGCTGACCCTCCT-3¢,pCAS2-RTR:5¢-TTGCTGAGAAGGCGTGGTAGAG-3¢)进行PCR扩增,PCR产物切胶回收纯化,进行Sanger测序。
1.6 小鼠组织RNA提取及反转录
采用断颈法处死实验小鼠,依次分离出晶状体、大脑、肝脏、脾脏、肺、肾脏组织,PBS冲洗后剪取0.1 g组织使用Trizol提取组织总RNA。分别取1 μg总RNA进行反转录,cDNA于–20℃冰箱保存备用。
1.7 PCR检测小鼠组织TSR1表达
利用PCR检测小鼠各组织表达情况,TSR1上游引物为:5¢-CGGTGTATTTGAGTGAACGGG-3¢,下游引物为:5¢-CAGATCCCCTGGTCTTGCAT-3¢,内参GAPDH上游引物为:5¢-AGGTCGGTGTGAACGGATTTG-3¢,下游引物为:5¢-GGGGTCGTTGATGGCAACA-3¢。反应条件为94℃预变性5 min,94℃变性30 s、60℃退火30 s、72℃延伸30 s,共30个循环,72℃延伸5 min。
1.8 人晶状体上皮细胞SRA01/04蛋白提取
利用RAPI裂解液提取细胞总蛋白,细胞浆蛋白和细胞核蛋白提取按照ProteinExtTMMammalian Nuclear and Cytoplasmic Protein Extraction Kit (DE201,北京全式金生物技术有限公司)说明书进行,于−80℃保存。
1.9 Western blotting检测TSR1蛋白表达
SRA01/04细胞总蛋白、细胞浆蛋白和细胞核蛋白12% SDS-PAGE电泳并转移至PVDF膜,5%脱脂奶粉室温封闭1 h,TSR1一抗(1∶1000) (ab220639, abcam, UK)和β-actin一抗(1∶1000) (KGAA001-1,江苏凯基生物技术股份有限公司,南京市)4℃孵育过夜,辣根过氧化物酶标记的二抗(KGAA35和KGAA37,江苏凯基生物技术股份有限公司)室温孵育,TBST洗涤后滴加显色液在化学发光凝胶检测系统中显影检测。
1.10 免疫荧光实验
年龄相关性白内障患者晶状体前囊膜组织手术取出后立刻包埋于组织包埋剂O.T.C中,冰冻切片厚度为5 μm,切片立刻置于预冷的丙酮中固定;24周人胎眼石蜡切片置于二甲苯Ⅰ和二甲苯Ⅱ中脱蜡处理,再依次置于体积分数100%、95%、90%、80%和70%的梯度酒精中浸泡。PBS冲洗后将切片浸泡于抗原修复液中进行抗原修复,封闭后滴加TSR1抗体(1∶200稀释)和ACTB抗体(1∶100稀释) 4℃孵育过夜。次日使用PBS冲洗后滴加两种二抗混合液避光孵育,DAPI染液(100 mg/mL)复染,封片剂封片,使用尼康A1R激光共聚焦显微镜观察拍照。
1.11 生物信息学分析
使用iSyTE数据库检索Tsr1和相关基因在不同时期的小鼠晶状体组织中的表达变化情况,使用在线软件DAVID进行GO分析和KEGG分析,使用bioGRID在线软件绘制蛋白质–蛋白质相互作用网络PPI图。
2 结果与分析
2.1 先天性白内障家系基因突变分析
本研究共收集了3代18人血样,其中患者8人,正常人10人(图1A)。全外显子组测序数据经筛选后,人群频率小于万分之一的杂合位点共14个(表1),候选位点在家系中进行Sanger测序验证,结果显示c.202-1G>A杂合突变在家系中与患者白内障表型共分离(图1B)。ExAC、dbSNP、EVS、GnomAD、1000genomes、CMDB和NovoDb等正常人数据库中均未见该变异位点,HGMD和ClinVar等疾病数据库也未见该位点的报道,家系正常人和105例正常对照不携带该变异位点。c.202-1G>A位点在物种间高度保守(图1C),Human splicing finder 3.1在线软件预测突变会影响外显子的正常剪接(图1D)。
2.2 Minigene实验检测突变对外显子剪接的影响
野生型和突变型minigene质粒转染SRA01/04细胞,24 h后提取总RNA进行RT-PCR,琼脂糖凝胶电泳结果表明pCAS2空载和野生型minigene可以正常剪接,野生型和突变型条带大小无明显差异(图2A)。Sanger测序表明突变型c.202-1G>A激活了外显子3的隐蔽剪接位点,导致突变型mRNA缺少一个碱基G,预测TSR1蛋白第68位氨基酸后发生移码,并于其后46位形成终止密码子,p.(Phe68Valfs*46) (图2B),与human splicing finder 3.1在线软件预测结果一致。
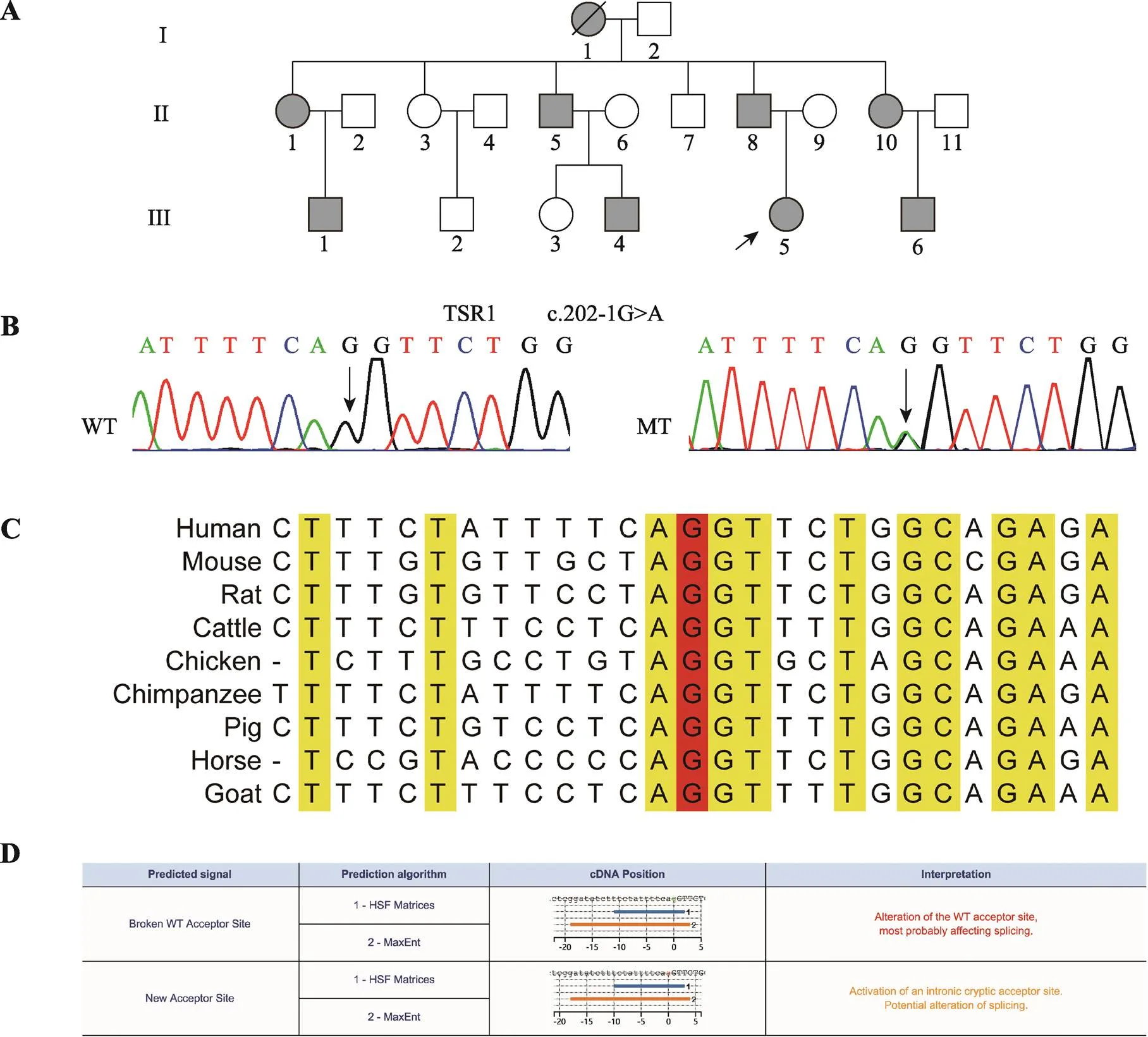
图1 先天性白内障家系TSR1变异分析
A:先天性白内障家系系谱图。实心圆圈表示女性患者,实心方块表示男性患者,空心圆圈表示女性正常人,空心方块表示男性正常人,黑色箭头表示先证者,黑色斜线表示个体已死亡。B:Sanger测序图。WT为野生型,MT为c.202-1G>A突变型,箭头所指为突变位点。C:位点保守性分析结果。红色G表示突变位点。D:Human splicing finder预测结果。突变破坏了正常受体位点,形成了一个新的剪接受体位点。
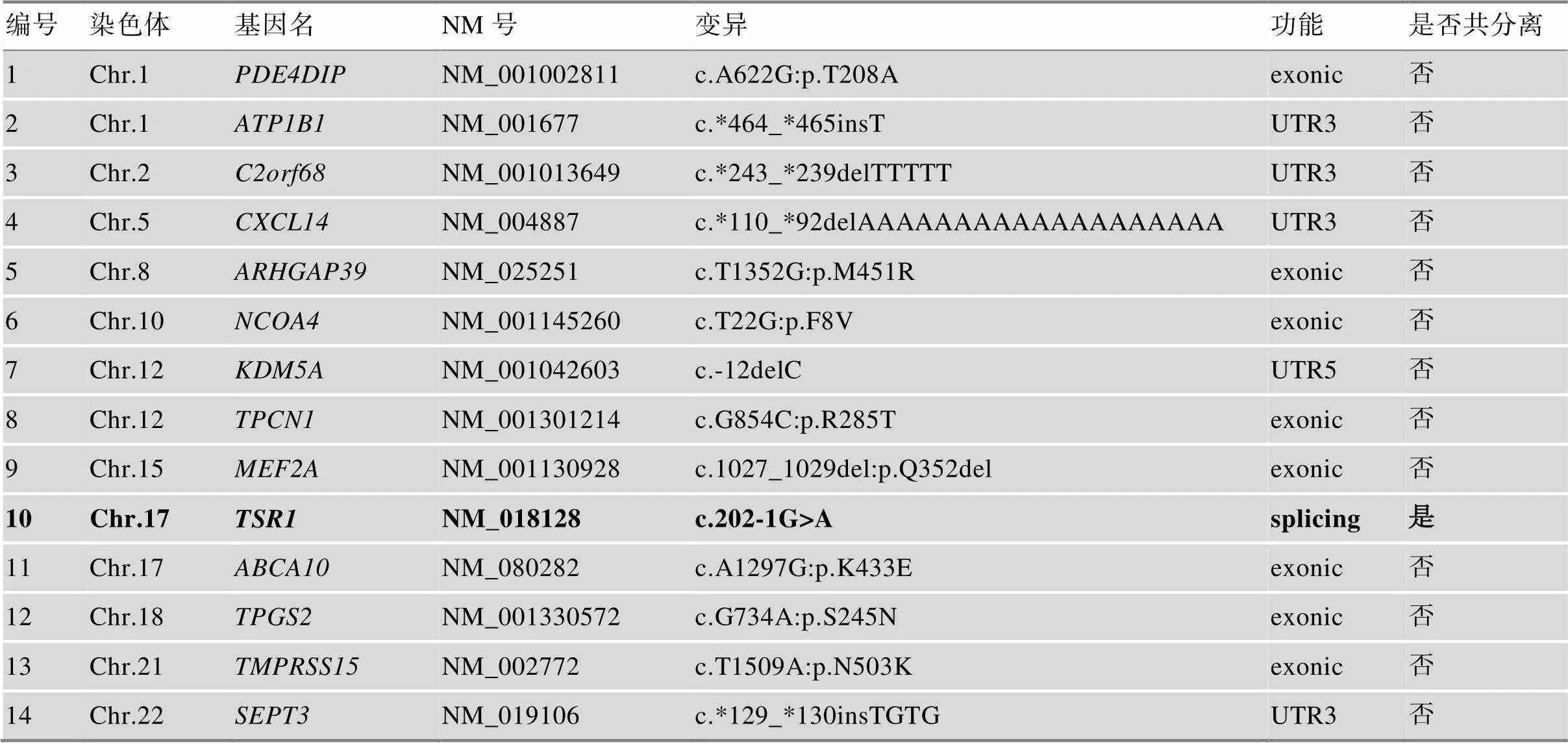
表1 全外显子组测序可疑位点Sanger测序验证情况
*表示终止密码子。
2.3 RT-PCR检测小鼠各组织Tsr1表达
以小鼠晶状体、大脑、肝脏、脾脏、肺和肾脏组织cDNA为模板进行RT-PCR,结果显示基因在所选取的6种小鼠组织中均有表达(图3),表达量并无明显差异。
2.4 Western blotting检测人晶状体上皮细胞SRA01/04中TSR1表达
Western blotting结果显示TSR1在人晶状体上皮细胞SRA01/04细胞核和细胞质中均有表达,且表达量都很高(图4)。
2.5 免疫荧光实验检测人晶状体中TSR1表达和定位
免疫荧光结果显示TSR1在年龄相关性白内障患者晶状体前囊膜组织细胞有表达,在细胞核和细胞质中均有表达(图5A)。在24周人胎眼晶状体纤维细胞、晶状体上皮细胞以及晶状体前囊组织中均有表达(图5B)。
2.6 生物信息学分析结果
在胚胎期和出生后的小鼠晶状体组织中持续表达(图6A),晶状体特异性CBP:p300双敲除的小鼠晶状体中表达下调(图6B)。使用iSyTE的Co-expression功能获取双敲除小鼠中与Tsr1表达模式相同的一组基因进行GO功能、KEGG信号通路分析及PPI分析,GO分析结果显示,与Tsr1表达模式相同的基因主要参与了代谢、生物调节、应激等生物过程。在分子功能方面,主要参与核糖体构成、金属离子结合等相关功能(图6C)。KEGG信号通路分析显示与Tsr1表达模式相同的基因在核糖体形成、氧化磷酸化、泛素介导的蛋白水解和亨廷顿舞蹈病4个通路中富集(图6D)。PPI结果显示基因被聚类到5个不同的信号通路上,其中Rps21、Rps29、Rpl38、Rplp2、Rpl31和Imp3与Tsr1存在直接相互作用(图6E)。
3 讨论
先天性白内障遗传异质性高,致病基因众多,致病机制复杂,同一基因突变在不同家系中的表型可能不同,即使在一个家系内,患者的表型也可能不尽相同。这也导致临床诊断和遗传学诊断困难重重。随着高通量测序技术的发展和人类基因组数据库的完善,已经有越来越多的先天性白内障致病基因和突变位点被发现[11~16]。
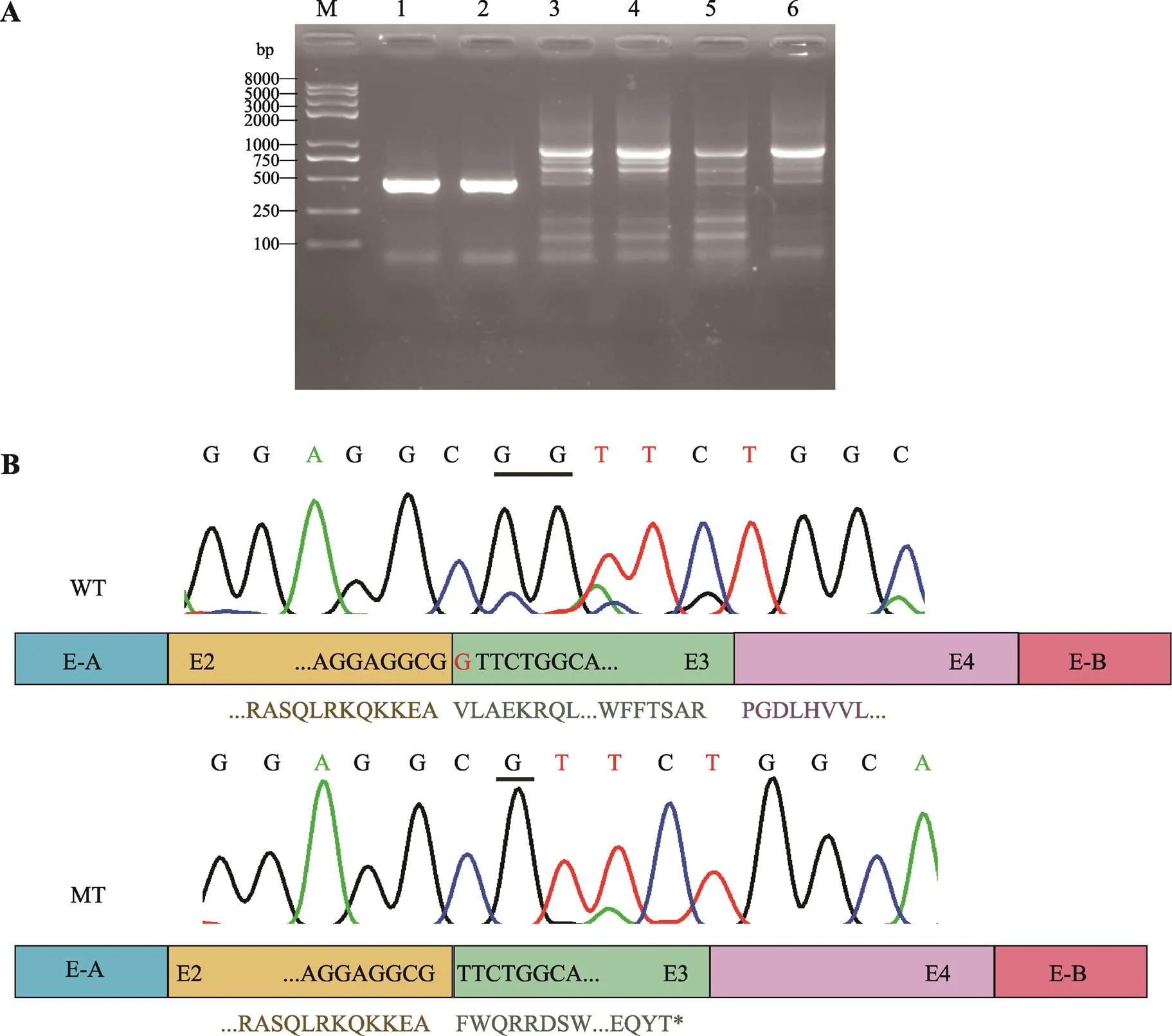
图2 Minigene分析
A:RT-PCR产物电泳结果。M:DNA Marker trans2k plusII;1:pCAS2空白载体;2:pCAS2空白载体;3:野生型pCAS2-TSR1质粒;4:野生型pCAS2-TSR1质粒;5:突变型pCAS2-TSR1c.202-1G>A质粒;6:突变型pCAS2-TSR1c.202-1G>A质粒。B:野生型和突变型RT-PCR产物Sanger测序结果。WT:野生型;MT:突变型。野生型可以正常转录,突变型产生缺失一个碱基“G”的转录本,导致TSR1蛋白移码。*形成终止密码子。
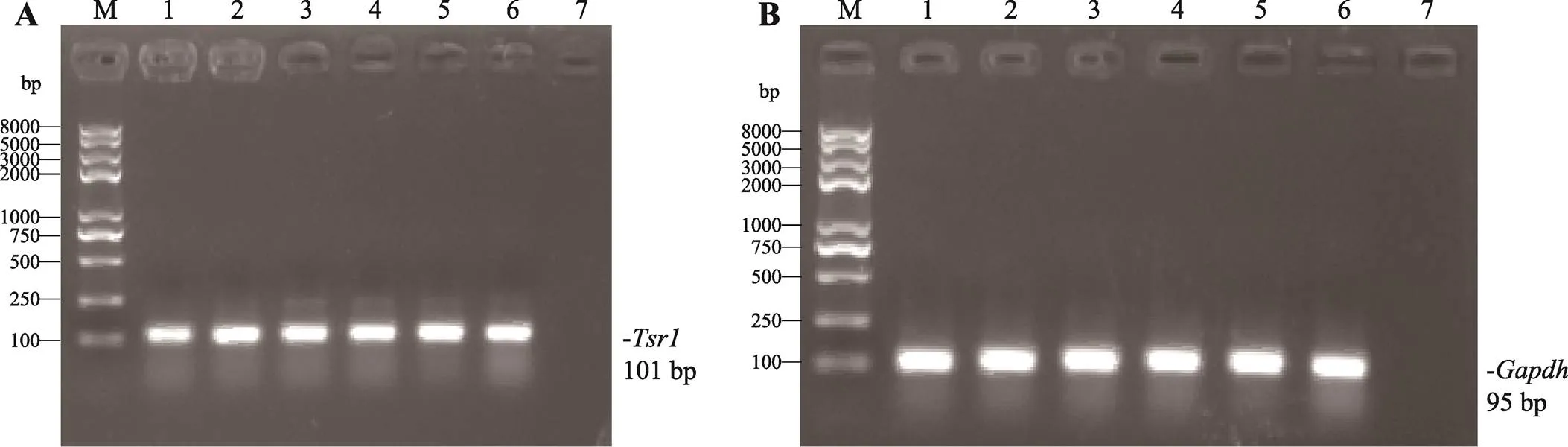
图3 Tsr1在小鼠不同组织中的表达
A:在小鼠各组织中的表达。B:在小鼠各组织中的表达。M:DNA Marker trans2k plusⅡ;1:晶状体;2:脑;3:肝;4:脾;5:肺;6:肾;7:空白对照。
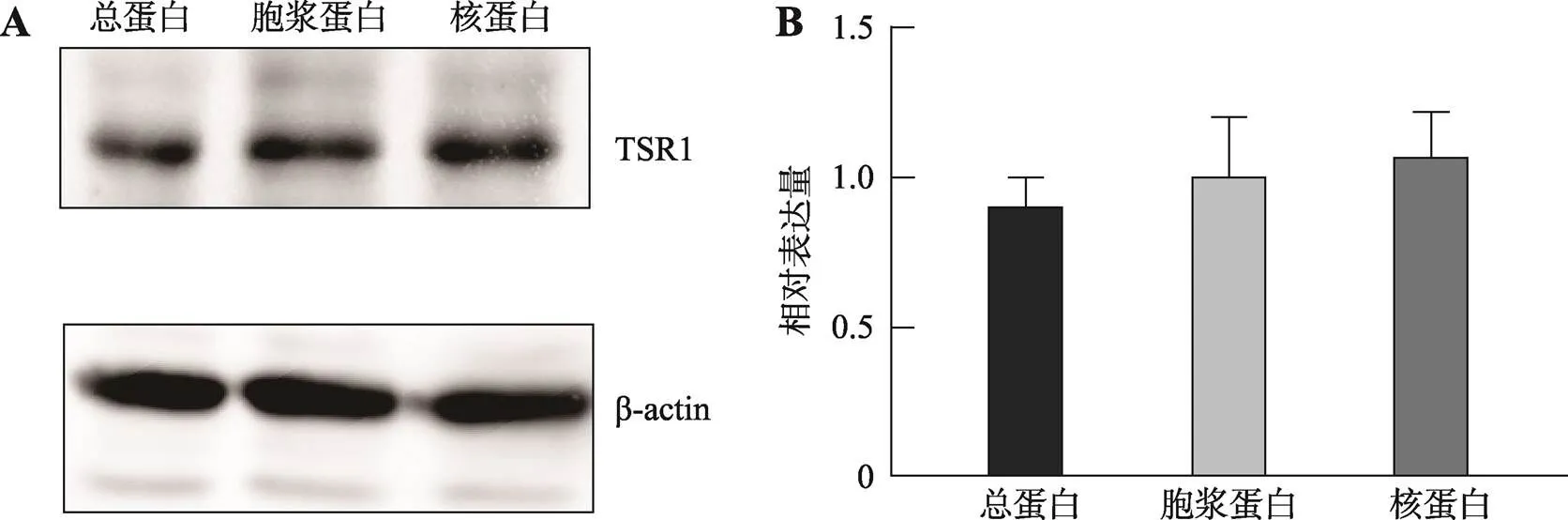
图4 TSR1在SRA01/04细胞中的表达
A:Western blotting检测TSR1在SRA01/04中的表达。;B:定量分析TSR1在SRA01/04中的表达。TSR1在细胞质和细胞核中均有表达,没有显著差异。
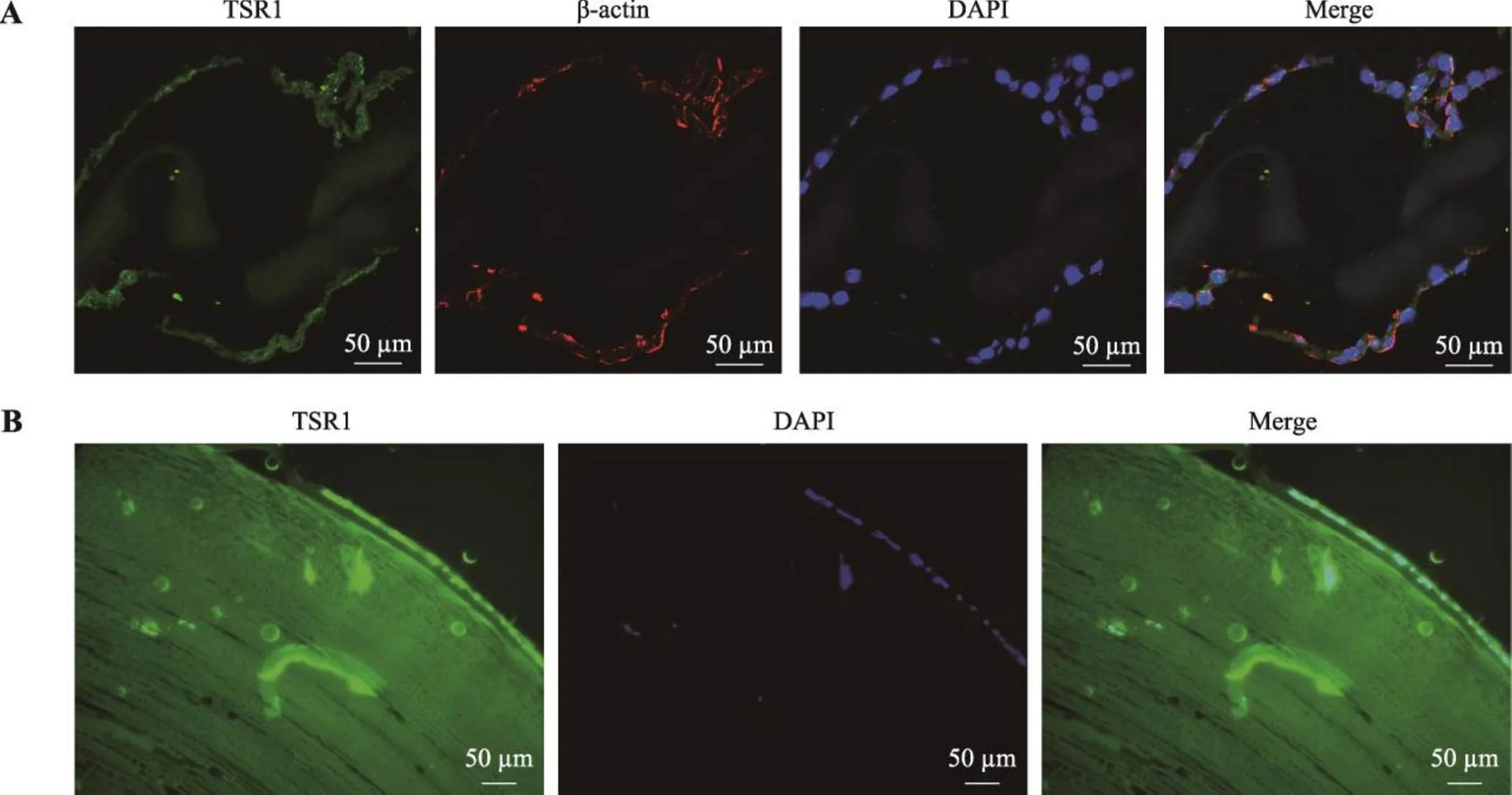
图5 免疫荧光法检测人晶状体TSR1的表达及定位
A:年龄相关性白内障患者晶状体前囊免疫荧光结果。绿色荧光:TSR1;红色荧光:β-actin;蓝色:DAPI。B:24周人胎眼晶状体组织免疫荧光结果。绿色荧光:TSR1;蓝色:DAPI。
先天性白内障是最早被发现呈常染色体显性遗传的人类疾病。近年来,国内多个研究小组利用中国人群的白内障家系资源优势,在致病基因定位克隆和突变基因鉴定方面取得了突出成绩。2002年,中国科学院上海生命科学研究院孔祥银团队发现编码热休克转录因子家族成员的基因突变可引起常染色体显性板层白内障和Marner白内障[17]。2005年,中国医学科学院沈岩团队报道基因突变导致皮质性白内障[18]。2009年,中国医学科学院张学团队发现基因突变导致后极性白内障[19]。2014年,青岛眼科医院谢立信团队报道基因突变导致白内障小角膜综合征[20]。2019年,中国人民解放军第三军医大学大坪医院叶剑团队报道基因突变导致后极性白内障[21]。
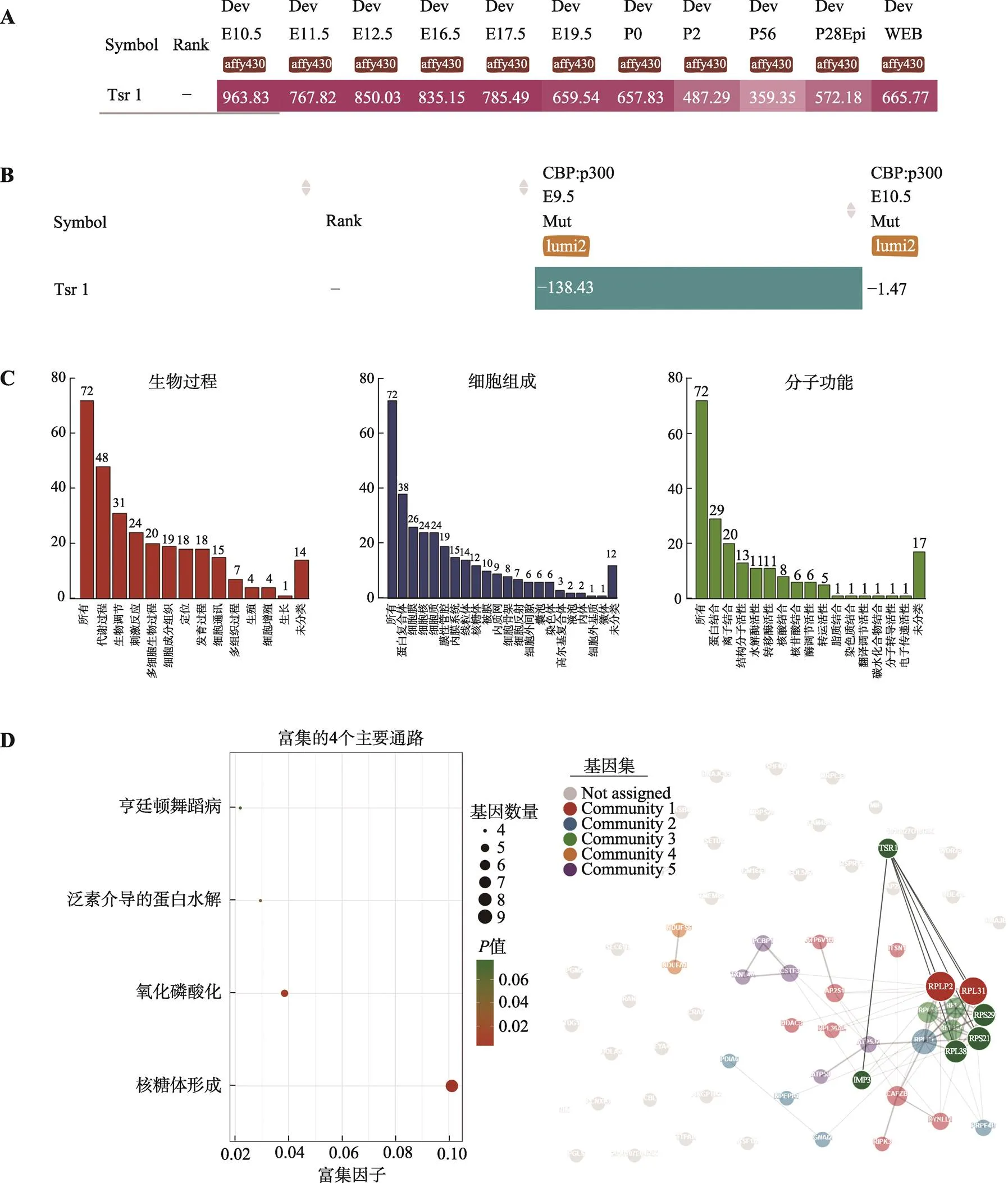
图6 TSR1表达模式、GO功能、KEGG信号通路及PPI等生物信息学分析
A:iSyTE数据库中表达模式。检索iSyTE数据库,发现Tsr1在小鼠晶状体发育各个阶段均持续表达。B:CBP: p300双敲除小鼠晶状体Tsr1表达。CBP: p300双敲除小鼠晶状体Tsr1表达下降。C:GO功能分析。提取与Tsr1表达模式相同的基因进行GO分析。D:KEGG分析。KEGG分析结果表明基因主要被富集到核糖体形成、氧化磷酸化、泛素介导的蛋白水解和亨廷顿舞蹈病4个通路。E:PPI分析。提取与Tsr1表达模式相同的基因进行蛋白–蛋白相互作用网络分析,发现表达下降的基因其编码蛋白Rps21、Rps29、Rpl38、Rplp2、Rpl31和Imp3与Tsr1直接相互作用。
本研究通过全基因组测序技术,在一个中国常染色体显性遗传先天性白内障家系检测到一个可疑的候选基因变异c.202-1G>A,p.(Phe68Valfs*46),该变异在家系中与白内障表型共分离,正常人数据库未见该位点的报道,疾病数据库中未见该基因与人类疾病的相关报道,105名正常对照个体未携带此突变,因此可能是一个新先天性白内障致病基因。
(NM_018128.4)位于17p13.3,含15个外显子,编码804个氨基酸。编码前体rRNA加工蛋白,在40S核糖体成熟过程中发挥重要作用[22]。基因c.202-1G位点高度保守,minigene实验证实该变异影响了基因mRNA剪接导致蛋白移码。RT-PCR、免疫荧光和Western blotting实验证实了TSR1在小鼠和人的晶状体组织和细胞系中均有表达。通过检索iSyTE数据库,本研究发现在胚胎期和出生后的小鼠晶状体组织中持续表达,且在CBP:p300双敲除的白内障小鼠晶状体中表达下调[23],推测其可能在晶状体的发育过程中发挥重要作用并参与了敲除小鼠白内障表型的形成。通过敲除小鼠晶状体中与Tsr1表达模式相同基因的蛋白质–蛋白质相互作用(PPI)分析发现,表达下调的基因中参与核糖体形成的Rps21、Rps29、Rps38、Rplp2、Rpl31和Imp3均与Tsr1有直接相互作用,其中Rps21、Rps29、Rps38、Rplp2和Rpl31均为核糖体蛋白,参与核糖体的组装和成熟[24~27]。目前已有报道核糖体蛋白L21、L15、L13a和L7a表达下调可以导致年龄相关性白内障,可能原因是核糖体异常导致的晶状体蛋白合成障碍[28]。由于没有匹配的相同年龄性别的正常人晶状体前囊膜组织做对照,本研究只检测了TSR1在年龄相关性白内障患者前囊膜组织有表达,没有进行定量分析其表达变化情况。在CBP:p300双敲除小鼠晶状体中,晶状体蛋白α、β和γ家族基因和多个晶状体发育的关键转录因子表达均有不同程度的下调[23],推测这些基因的表达下调可能是由核糖体的组装异常引起的。TSR1作为核糖体组装的重要成分之一,可能与其他表达下调的核糖体蛋白一同参与双敲除小鼠晶状体表型形成。在表达下调的基因中,与Tsr1存在直接相互作用的Imp3蛋白参与MAPK-Erk信号通路,Imp3通过失活衔接蛋白Ksr1进而抑制MAPK-Erk通路[29]。在晶状体中,MAPK信号通路的增强可以诱导晶状体上皮细胞的凋亡,进而抑制白内障的发生[30]。当Imp3基因下调时,Ksr1对MAPK通路的抑制作用增强,晶状体上皮细胞凋亡减少,细胞过量增殖和迁移,最终可能导致白内障的发生。Tsr1与Imp3的相互作用机制尚不明确,Tsr1是否能与Imp3一同调节MAPK通路,进而影响晶状体细胞的凋亡和增殖还需要进一步研究确定。
综上所述,本研究在一个中国先天性白内障家系中通过全基因组测序及Sanger测序验证,检测到变异c.202-1G>A,p.(Phe68Valfs*46),与家系疾病表型共分离。通过minigene实验证实该变异影响了基因mRNA剪接。Western blotting、免疫荧光和RT-PCR实验证实了TSR1在人晶状体上皮细胞SRA01/04、年龄相关性白内障患者晶状体前囊膜组织、24周人胎眼晶状体和小鼠晶状体中表达。利用iSyTE、GO分析、KEGG分析和PPI数据库分析,为进一步明确TSR1在晶状体中的功能提供了有价值的研究线索。
[1] Foster A, Gilbert C, Rahi J. Epidemiology of cataract in childhood: a global perspective., 1997, 23(Suppl. 1): 601–604.
[2] Gilbert C, Foster A. Childhood blindness in the context of VISION 2020--the right to sight., 2001, 79(3): 227–232.
[3] Rahi JS, Sripathi S, Gilbert CE, Foster A.Childhood blindness in India: causes in 1318 blind school students in nine states.(Lond), 1995, 9(Pt 5): 545–550.
[4] Tai F, Yu L, Zhao HX, Wang ML. Report of a Big Family of Marinesco-Sjögren Syndrome in 11 Case of Five Generation with X-Linked Recessive Inheritance., 2001, 23(5): 409–741.邰风, 于丽, 赵洪祥, 王明礼. X连锁隐性遗传性Marinesco-Sjögren综合征一家系五代11例报告. 遗传, 2001, 23(5): 409–741.
[5] Wang F, Chen D, Jiao T, Yang HQ. New mutation of GJA8 gene in a family with congenital cataract in northern China., 2019, 39(9): 854–856.王凡, 陈迪, 焦婷, 杨会琴. 中国北方一先天性白内障家系GJA8基因新突变. 眼科新进展, 2019, 39(9): 854– 856.
[6] Hu SS, Ma Z, Zhao BW, Lin H, Qi YH. Mechanism of mutation of lens major intrinsic protein causing congenital cataract., 2015, 35(3): 218–222.胡姗姗, 马笮, 赵保文, 林辉, 齐艳华. 晶状体主要内源性蛋白MIP的突变体致先天性白内障的发生机制. 眼科新进展, 2015, 35(3): 218–222.
[7] Imagawa E, Fukai R, Behnam M, Goyal M, Nouri N, Nakashima M, Tsurusaki Y, Saitsu H, Salehi M, Kapoor S, Tanaka F, Miyake N, Matsumoto N.Two novel homozygous RAB3GAP1 mutations cause warburg micro syndrome., 2015, 2: 15034.
[8] Yamashita T, Mitsui J, Shimozawa N, Takashima S, Umemura H, Sato K, Takemoto M, Hishikawa N, Ohta Y, Matsukawa T, Ishiura H, Yoshimura J, Doi K, Morishita S, Tsuji S, Abe K.Ataxic form of autosomal recessive PEX10- related peroxisome biogenesis disorders with a novel compound heterozygous gene mutation and characteristic clinical phenotype., 2017, 375: 424–429.
[9] De Franco E, Flanagan SE, Yagi T, Abreu D, Mahadevan J, Johnson MB, Jones G, Acosta F, Mulaudzi M, Lek N, Oh V, Petz O, Caswell R, Ellard S, Urano F, Hattersley AT.Dominant ER stress-inducing WFS1 mutations underlie a genetic syndrome of neonatal/infancy-onset diabetes, congenital sensorineural deafness, and congenital cataracts., 2017, 66(7): 2044–2053.
[10] Huang KM, Wu JH, Brooks SP, Hardcastle AJ, Lewis RA, Stambolian D. Identification of three novel NHS mutations in families with Nance-Horan syndrome., 2007, 13: 470–474.
[11] Ma AS, Grigg JR, Ho G, Prokudin I, Farnsworth E, Holman K, Cheng A, Billson FA, Martin F, Fraser C, Mowat D, Smith J, Christodoulou J, Flaherty M, Bennetts B, Jamieson RV. Familial congenital cataracts: mutational Spectrum and new diagnoses using next-generation sequencing., 2016, 37(4): 371–384.
[12] Patel N, Anand D, Monies D, Maddirevula S, Khan AO, Algoufi T, Alowain M, Faqeih E, Alshammari M, Qudair A, Alsharif H, Aljubran F, Alsaif HS, Ibrahim N, Abdulwahab FM, Hashem M, Alsedairy H, Aldahmesh MA, Lachke SA, Alkuraya FS.Novel phenotypes and loci identified through clinical genomics approaches to pediatric cataract., 2017, 136(2): 205–225.
[13] Javadiyan S, Craig JE, Souzeau E, Sharma S, Lower KM, Mackey DA, Staffieri SE, Elder JE, Taranath D, Straga T, Black J, Pater J, Casey T, Hewitt AW, Burdon KP. High-throughput genetic screening of 51 pediatric cataract genes identifies causative mutations in inherited pediatric cataract in south eastern Australia., 2017, 7(10): 3257–3268.
[14] Gillespie RL, O'Sullivan J, Ashworth J, Bhaskar S, Williams S, Biswas S, Kehdi E, Ramsden SC, Clayton- Smith J, Black GC, Lloyd IC. Personalized diagnosis and management of congenital cataract by next-generation sequencing., 2014, 121(11): 2124–2137.
[15] Liu YQ, Zhu X, Li SJ, Yang YM, Yang Mu, Zhao PQ, Zhu XJ. Identification of LRP5 mutations in families with familial exudative vitreoretinopathy., 2017, 39(3): 241–249.刘玉庆, 朱雄, 李姝锦, 杨业明, 杨牧, 赵培泉, 朱献军. 家族性渗出性玻璃体视网膜病变患者LRP5基因突变研究. 遗传, 2017, 39(3): 241–249.
[16] Yang XL, Su YH, Li WL. Progress in animal models for inherited cataract., 2007, 29(2): 137– 144.杨秀兰,苏玉虹,李文龙. 遗传性白内障动物模型的研究进展. 遗传, 2007, 29(2): 137–144.
[17] Bu L, Jin YP, Shi YF, Chu RY, Ban AR, Eiberg H, Andres L, Jiang HS, Zheng GR, Qian MQ, Cui B, Xia Y, Liu J, Hu LD, Zhao GP, Hayden MR, Kong XY. Mutant DNA- binding domain of HSF4 is associated with autosomal dominant lamellar and Marner cataract., 2002, 31(3): 276–278.
[18] Sun H, Ma Z, Li Y, Liu B, Li Z, Ding X, Gao Y, Ma W, Tang X, Li X, Shen Y. Gamma-S crystallin gene (CRYGS) mutation causes dominant progressive cortical cataract in humans., 2005, 42(9): 706–710.
[19] Zhang TX, Hua R, Xiao W, Burdon KP, Bhattacharya SS, Craig JE, Shang DD, Zhao XL, Mackey DA, Moore AT, Luo Y, Zhang JS, Zhang X. Mutations of the EPHA2 receptor tyrosine kinase gene cause autosomal dominant congenital cataract., 2009, 30(5): E603–E611.
[20] Chen P, Dai YH, Wu XM, Wang Y, Sun SY, Xiao JJ, Zhang QY, Guan LP, Zhao XW, Hao XD, Wu RH, Xie LX. Mutations in the ABCA3 gene are associated with cataract-microcornea syndrome., 2014, 55(12): 8031–8043.
[21] Sun M, Chen C, Hou S, Li X, Wang H, Zhou J, Chen X, Liu P, Kijlstra A, Lin S, Ye J. A novel mutation of PANK4 causes autosomal dominant congenital posterior cataract., 2019, 40(4): 380–391.
[22] Scaiola A, Peña C, Weisser M, Böhringer D, Leibundgut M, Klingauf-Nerurkar P, Gerhardy S, Panse VG, Ban N. Structure of a eukaryotic cytoplasmic pre-40S ribosomal subunit., 2018, 37(7): e98499.
[23] Wolf L, Harrison W, Huang J, Xie Q, Xiao NN, Sun J, Kong LK, Lachke SA, Kuracha MR, Govindarajan V, Brindle PK, Ashery-Padan R, Beebe DC, Overbeek PA, Cvekl A. Histone posttranslational modifications and cell fate determination: lens induction requires the lysine acetyltransferases CBP and p300., 2013, 41(22): 10199–10214.
[24] Farrar JE, Quarello P, Fisher R, O'Brien KA, Aspesi A, Parrella S, Henson AL, Seidel NE, Atsidaftos E, Prakash S, Bari S, Garelli E, Arceci RJ, Dianzani I, Ramenghi U, Vlachos A, Lipton JM, Bodine DM, Ellis SR. Exploiting pre-rRNA processing in Diamond Blackfan anemia gene discovery and diagnosis., 2014, 89(10): 985–991.
[25] Martinez-Azorin F, Remacha M, Ballesta JP. Functional characterization of ribosomal P1/P2 proteins in human cells., 2008, 413(3): 527–534.
[26] Mirabello L, Macari ER, Jessop L, Ellis SR, Myers T, Giri N, Taylor AM, McGrath KE, Humphries JM, Ballew BJ, Yeager M, Boland JF, He J, Hicks BD, Burdett L, Alter BP, Zon L, Savage SA. Whole-exome sequencing and functional studies identify RPS29 as a novel gene mutated in multicase Diamond-Blackfan anemia families., 2014, 124(1): 24–32.
[27] Arthurs C, Murtaza BN, Thomson C, Dickens K, Henrique R, Patel HRH, Beltran M, Millar M, Thrasivoulou C, Ahmed A. Expression of ribosomal proteins in normal and cancerous human prostate tissue., 2017, 12(10): e0186047.
[28] Zhang WY, Hawse J, Huang QL, Sheets N, Miller KM, Horwitz J, Kantorow M. Decreased expression of ribosomal proteins in human age-related cataract., 2002, 43(1): 198–204.
[29] Hayes SD, Liu H, MacDonald E, Sanderson CM, Coulson JM, Clague MJ, Urbé S. Direct and indirect control of mitogen-activated protein kinase pathway-associated components, BRAP/IMP E3 ubiquitin ligase and CRAF/ RAF1 kinase, by the deubiquitylating enzyme USP15., 2012, 287(51): 43007–43018.
[30] Zhang Y, Huang WR. Sanguinarine induces apoptosis of human lens epithelial cells by increasing reactive oxygen species via the MAPK signaling pathway., 2019, 19(5): 4449–4456.
Studies of congenital cataract-relatedmutation and its expression in the lens
Yajie Yu1,2, Feng Qiu3, Xin-an Zhang1
Congenital cataract (CC) is a rare disease with dysplasia of the lens, mainly characterized by partial or complete opacity of the lens. The molecular basis of the disease is complex, mutations in over 266 genes associated with congenital cataracts had been reported. In this study, a novel congenital cataract candidate genewas identified by whole genome sequencing and Sanger sequencing in a Chinese congenital cataract family. Thec.202-1G>A substitution affected splicing ofmRNA was confirmed by a minigene assay. The expression ofin mouse lens, anterior lens capsule of age-related cataract patients and 24-week human fetal lens were determined by RT-PCR, Western blotting, and immunofluorescence assays. The expression ofin the embryonic and different developmental stages of the mouse lens was confirmed by analyzing the iSyTE database. The expression ofwas down-regulated in the lens-specific CBP:p300 double knockout mouse, and a set of genes with the same expression pattern ofin the CBP:p300 double knockout mouse lens were extracted for protein-protein interaction network analysis, and six proteins were screened for direct interaction with Tsr1. GO function analysis indicated thatmight play a role in the MAPK-Erk signaling pathway in addition to its involvement in ribosome assembly. This study provided valuable research clues to further clarify the function ofin the lens.
TSR1; lens; protein-protein interaction
2019-10-14;
2020-01-01
国家自然科学基金项目(编号:81572243)资助[Supported by the National Natural Science Foundation of China (No. 81572243)]
于雅洁,硕士研究生,专业方向:遗传学。E-mail: 907307673@qq.com
张新安,博士,教授,研究方向:基础医学。E-mail: zhangxa2725@163.com
10.16288/j.yczz.19-166
2020/1/8 14:48:26
URI: http://kns.cnki.net/kcms/detail/11.1913.r.20200107.1722.002.html
(责任编委: 徐湘民)
猜你喜欢
内蒙古民族大学学报(自然科学版)(2022年2期)2022-11-22 06:44:43
电子科技大学学报(2022年5期)2022-10-29 01:57:52
中国实用医药(2021年7期)2021-04-16 11:21:38
中国生殖健康(2020年4期)2021-01-18 02:58:10
肿瘤防治研究(2020年5期)2020-07-09 13:06:08
生物学教学(2019年9期)2019-09-23 03:53:02
中医眼耳鼻喉杂志(2019年2期)2019-04-13 05:23:52
中国生殖健康(2018年4期)2018-11-06 07:12:16
中外医疗(2015年5期)2015-08-29 01:54:42
西南国防医药(2015年11期)2015-02-28 19:38:55
