
牙鲆甲状腺激素受体TRαA介导甲状腺激素调控的靶基因鉴定
2020-04-06 05:09付元帅施志仪季文瑶陶家康
水生生物学报 2020年2期
谢 燕 付元帅 施志仪 季文瑶 陶家康
(上海海洋大学农业部淡水水产种质资源重点实验室, 上海 201306)
牙鲆Paralichthys olivaceusTemminck & Schlegel属于鲽形目, 在仔鱼向稚鱼的胚后发育中经历剧烈的变态过程, 表现为右眼左移、体位从侧卧变为平卧、浮游转为底栖生活等。甲状腺激素(Thyroid Hormone, TH)在鲽形目鱼类变态过程中起着关键性的调控作用[1—3], 直接控制着仔鱼变态的进程。T3(3, 3′, 5-triiodo-L-thyronine,三碘甲状腺原氨酸)是TH发挥生理作用的主要形式。TH是通过甲状腺激素受体(Thyroid Hormone Receptors, TRs)和非甲状腺激素受体发挥作用[4,5]。甲状腺激素受体属于核受体超家族中的一员, 是配体T3诱导的转录因子, 在介导T3的作用过程中处于核心地位, 通过招募多种共激活子或共抑制子来实现基因转录的激活或抑制。TRs分子在结构上分为6个区, 从氨基端到羧基端依次为A-F区, 组成3个功能域: 转录激活域AF-1(A/B区), DNA结合域DBD(C区), 铰链区(D区), 配体结合域LBD(E区)[6,7]。TRs与维甲酸X受体(Retinoid X receptors)及其他核受体形成异二聚体结合到靶基因启动子区域的甲状腺激素应答元件上(Thyroid Hormone Response Elements,TREs), 从而调控靶基因转录[8,9]。TREs通常包含两个或更多个串联排列的六聚体半位序列AGGT(C/A)A[10], 具有3种识别类型[11]: 回文结构(Palindrome, AGGTCATGACCT, 如生长激素Ⅰ即Gh1)、直接重复(Direct Repeat, 最常见DR4类型, AGGTCANNNNAGGTCA, 如kruppel-like因子9即Klf9)和倒置回文结构(Everted Repeat, 最常见6 bp间隔,TGACCTNNNNNNAGGTCA, 如髓磷脂碱性蛋白即myelin basic protein, Mbp)。牙鲆含有TRαA、TRαB和TRβ三种受体, TR亚型(TRαA、TRαB和TRβ1、TRβ2)在牙鲆变态期间基因表达具有时间特异性和组织特异性:TRαA在变态高峰期急剧增加,高峰期后又迅速下降;TRαB在仔鱼发育整个过程中都很低;TRβs的表达水平在变态高峰增加, 于变态高峰后达到峰值, 高水平的TRβs在变态完成的稚鱼中尚在持续[3,12,13]。
那么TH是如何调控牙鲆仔鱼的变态? 鉴定出TRs直接调控的靶基因是一个关键。通过牙鲆转录组及基因组TRE数据库筛选, 结合生物信息学分析,初步确定候选靶基因atoh8(Gene ID:109627718)。atoh8(Atonal homolog 8, atonal bHLH transcription factor 8), 属于碱性螺旋-环-螺旋(basic Helix-Loop-Helix, bHLH)转录因子, 参与斑马鱼视网膜细胞和体节肌肉细胞的发育分化, 在视网膜和体节发育的调控过程中,atoh8起着不可或缺的重要作用[14]。关于atoh8与甲状腺激素调控通路的关系尚未见报道。
本研究克隆了牙鲆TRαA基因及atoh8基因5′-侧翼序列各缺失片段, 并成功构建p3×Flag-TRαA和pGL3-Pro-atoh8-1517/1333/708(含候选靶基因5′-侧翼序列的缺失片段), 通过在HEK293T细胞中共转染p3×Flag-TRαA和pGL3-Pro-atoh8-1517/1333/708的双荧光素酶报告实验, 分别探究不同的5′-侧翼序列长度和对同一5′-侧翼序列进行不同处理(TRαA和T3都不加、只加TRαA或同时加TRαA与T3)对启动子转录活性的影响, 以期鉴定出TRαA受体是否结合在atoh8基因5′调控区特异的TRE序列来调控该基因的转录并判别具体是哪个TRE位点起到了关键作用, 最终鉴定出atoh8是否为TRαA介导甲状腺激素调控的直接靶基因。
1 材料与方法
1.1 材料与试剂
出膜后20d (20dph,days post hatching)、28d(28dph)的牙鲆仔鱼, 采自中国水产科学研究院北戴河中心实验站; HEK293T细胞购自ATCC细胞库。DMEM、胎牛血清、青霉素-链霉素、0.25%胰酶-EDTA购自Gibco; 细胞裂解液、蛋白酶抑制剂PMSF、ECL化学发光试剂盒购自上海威奥; 一抗Anti-Flag Tag (1E6) Monoclonal Antibody (GNI4110-FG)购自上海睿星, 二抗HRP-conjugated Goat Anti-Mouse IgG (D110087)、5×蛋白质加样缓冲液、平端 DNA 片段添dA试剂盒购自上海生工, 引物合成由上海生工完成; BCA蛋白定量试剂盒购自康为世纪, Anti-DYKDDDDK G1 Affinity Resin和DYKDDDDK Peptide购自金斯瑞; DNaseI、反转录试剂盒、FuGENE®HD转染试剂均购于Promega; 双荧光素酶报告基因检测试剂盒购自YEASEN,EcoRⅠ、KpnⅠ-HF、XhoⅠ限制酶、T4 DNA Ligase购自NEB; X-gal、IPTG购自天根, 2 × Phanta Max Master Mix购自Vazyme, pEASY®-T5 Zero购自全式金,DH5α购自上海唯地; TaKaRa LATaq、pMD19-T、TB GreenTMPremix ExTaqTMⅡ (Tli RNaseH Plus)购自TaKaRa, 琼脂糖凝胶DNA回收试剂盒、质粒小提、无内毒素质粒大提试剂盒购自Omega。p3×Flag、pGL3-Basic载体由上海海洋大学李名友教授惠赠。
1.2 方法
总RNA提取和反转录将28dph仔鱼按照Trizol®Reagent(Invitrogen)试剂盒说明书提取总RNA。检测总RNA纯度、浓度和完整性[15], 均符合要求后置于-80℃保存; 将上述RNA用DNaseⅠ处理后, 按Su等[16]方法反转录, 产物cDNA用于PCR扩增或-20℃保存。
牙鲆TRαA基因CDS区的克隆从NCBI(https://www.ncbi.nlm.nih.gov/)数据库中获取牙鲆TRαA基因序列(Gene ID: 109641414), 根据其序列和p3×Flag多克隆位点设计一对引物TRαA1252-F/R(表 1), 以上述cDNA为模板, 通过2×Phanta Max Master Mix扩增CDS区全长。
扩增产物经1%琼脂糖凝胶电泳, 胶回收纯化目的片段, 加A, 连接至pEASY®-T5 Zero, 转化DH5α。挑取阳性单克隆, 经菌液PCR验证有大小正确的片段插入后测序, 交由上海生工完成。
重组真核表达载体p3×Flag-TRαA的构建将上述测序正确的菌液和p3×Flag扩大培养提质粒;用EcoRⅠ、KpnⅠ37℃双酶切; 将鉴定正确的酶切PCR产物与p3×Flag载体分别进行胶回收, 产物按摩尔比3∶1经T4 DNA Ligase 16℃连接16h; 转化DH5α, 挑取阳性单克隆, 摇菌提质粒, 双酶切验证,再送上海生工测序验证; 将鉴定正确的p3×Flag-TRαA扩大培养, 提取不含内毒素的重组质粒。
HEK293T细胞培养及瞬时转染HEK293T细胞培养于10% FBS-DMEM 培养液中, 置于37℃,5% CO2的培养箱中常规培养, 转染前一天接种于6孔细胞培养板1.5 mL不含抗生素的10% FBSDMEM中, 转染时细胞密度为90%—95%。配制转染复合物[3 μg p3×Flag-TRαA(实验组)或p3×Flag(阴性对照), 7.5 μL FuGENE®HD转染试剂, 无抗生素、无血清DMEM补至500 μL], 混匀后室温孵育10—15min, 加到6孔板相应孔中, 未转染质粒作为空白对照, 每组(未转、空载、重组质粒)设4个重复, 一组用于总蛋白提取, 其余3组用于总RNA提取。培养6h左右换完全培养基, 48h后进行总蛋白和总RNA提取。
TRαA基因的RT-PCR及实时定量PCR检测将上文中RNA反转录成cDNA, PCR扩增TRαA基因CDS区全长与18S定量内参基因。在CFX96 TouchTMReal-Time PCR Detection System (Bio-Rad, USA)上分别制作目的基因TRαA和内参基因18S的标准曲线, 在符合要求后, 随后定量分析所有样品。反应体系(20 μL): 1 μL cDNA, 各1 μLTRαA-qF/R或18S-qF/R (表 1), 10 μL TB GreenTMPremix ExTaqTMⅡ(Tli RNaseH Plus)和7.0 μL DEPC水; 反应条件:95℃ 3min; 95℃ 10s, 60℃ 30s, 采集荧光40次。该实验中, 生物学重复n=3, 技术重复2次。
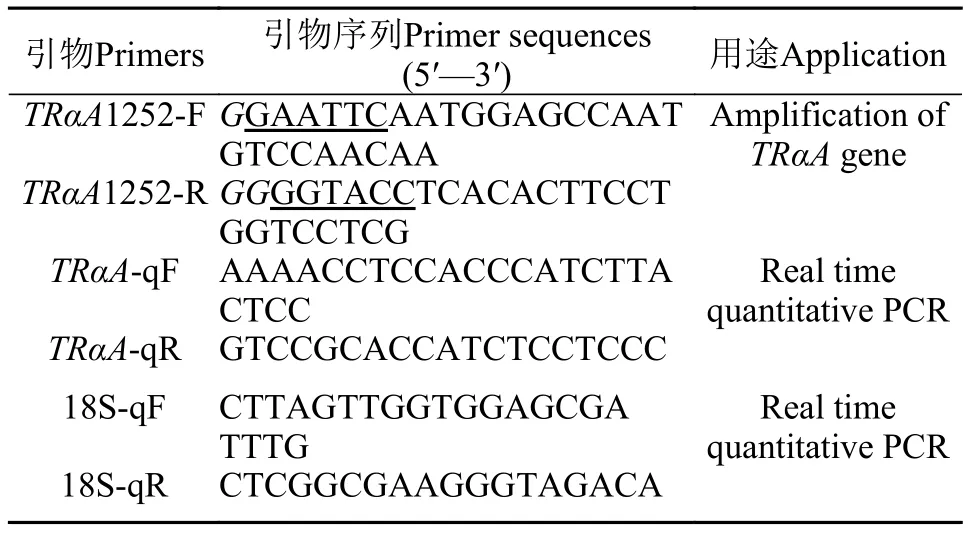
表 1 TRαA基因的扩增和定量引物Tab. 1 The primer used for cloning and real time quantitative PCR of TRαA gene
TRαA的mRNA相对表达量通过2-ΔΔCt[17]方法计算, 其结果以平均值±标准误差(Mean±SE)来表示(n=3)。通过SigmaPlot 12.0软件作图分析,利用SPSS 22软件中的One-Way ANOVA方差分析法分析不同样品间的相对表达差异, *P<0.05时表示差异显著。
Western blot检测融合蛋白的表达按上文方法转染HEK293T细胞, 经PBS洗2次, 1000 r/min离心5min分类收集HEK293T细胞; 加入预冷含PMSF (裂解液∶PMSF=99∶1)的细胞裂解液, 现配现用, 混匀后冰上轻微摇晃10min; 4℃、13000 r/min离心5min, 取上清, 分装使用或-80℃冻存。参考说明书用BCA试剂盒进行蛋白浓度测定。
分别吸取上述蛋白样品10 μg, 裂解液补至20 μL,加入5×SDS-PAGE加样缓冲液5 μL 100℃干式恒温10min, -20℃保存或加样进行SDS-PAGE电泳, 将分离的蛋白条带电转印至 PVDF 膜上, TBST洗2次(每次10min, 下述一样); 再将 PVDF 膜浸于含5%脱脂奶粉的TBS中室温封闭1h, TBST洗2次; 按1∶1000用TBS稀释一抗(小鼠抗Flag标签单抗)4℃孵育过夜, TBST洗3次; 按1∶5000用TBST稀释二抗(HRP标记的山羊抗小鼠 IgG)37℃孵育1h, TBST洗3次; 将PVDF膜与ECL发光液避光充分接触1min, 在暗室X光片下曝光拍照。
融合蛋白3×Flag-TRαA的纯化采用T25瓶整瓶细胞转染, 且质粒与转染试剂比为4 μg∶8 μL,按上述方法收集细胞提取总蛋白。目的蛋白TRαA的N端融合有3×Flag标签, 通过G1亲和层析柱纯化后, 分别保留总蛋白原液、流出液、洗杂液和洗脱液, 洗脱液经无菌滤器过滤除菌后, -80℃保存,Western blot检测纯化效果。
TRαA直接调控候选靶基因的筛选与重组报告基因表达载体pGL3-Pro-atoh8的构建 通过转录组数据(正常样品, 甲状腺素处理样品)筛查, 及基于现有上传的牙鲆基因组建立的TRE数据库筛选, 结合TREs识别类型, 从NCBI中获取牙鲆候选靶启动子5′-侧翼序列, 利用在线网站http://www.fruitfly.org/seq_tools/promoter.html, http://www.cbs.dtu.dk/services/Promoter/, https://www-bimas.cit.nih.gov/molbio/proscan/, http://biogrid-lasagna.engr.uconn.edu/lasagna_search/等预测靶启动子转录活性及转录因子结合位点。初步确定候选靶启动子, 如atoh8(Gene ID:109627718)。
采用苯酚抽提法从20dph仔鱼中提取基因组DNA。根据atoh8基因5′-侧翼序列和pGL3-Basic多克隆位点设计3对引物(表 2), 固定下游引物, 改变上游引物以缺失预测出的TRE位点, 以基因组DNA为模板, 经TaKaRa LATaq扩增先得到atoh8启动子最长的缺失片段。
按上述方法(产物连至pMD19-T, 用KpnⅠ、XhoⅠ双酶切, 酶切PCR产物和pGL3载体4℃连接过夜)获得pGL3-Pro-atoh8-1517。再以pGL3-Proatoh8-1517为模板, 按如上方法获得pGL3-Proatoh8-1333/708。
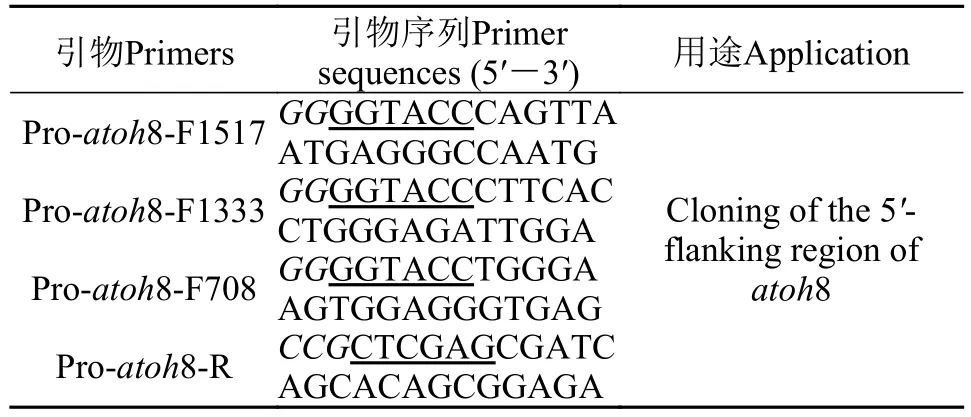
表 2 候选靶启动子各缺失片段的扩增引物Tab. 2 The primer used for cloning each missing fragment of the candidate target promoter
双荧光素酶报告实验参考前文方法将HEK293T细胞接种于96孔细胞培养板, 配制转染复合物[10 ng内参质粒 phRL-TK, 100 ng空载pGL3-Basic或100 ng重组质粒pGL3-Pro-atoh8-1517/1333/708(或含100 ng重组质粒p3×Flag-TRαA),0.22 μL FuGENE®HD 转染试剂, 无抗生素、无血清DMEM补至10 μL], 混匀后室温孵育10—15min,加到96孔细胞培养板相应孔中, 继续培养, 6h后换完全培养基, 再 24h后向转染的细胞中加入0.1 μL T3(终浓度为150 ng/μL), 再24h后检测并计算萤火虫荧光素酶与海肾荧光素酶的比值, 每个转染组设置3个复孔, 重复3次。数据采用SigmaPlot 12.0作图, 并通过SPSS 22软件中的单因素方差分析“一般线性模型”-“单变量”和LSD进行相对表达分析, 当*P<0.05时接受差异显著。
2 结果
2.1 牙鲆TRαA基因CDS区的克隆和p3×Flag-TRαA的构建
以28dph牙鲆仔鱼 cDNA 为模板进行PCR扩增,电泳结果显示片段大小约为1.2 kb, 与NCBI上传(XM_020105869.1)的大小相一致; 实际扩增得到片段大小是1252 bp, 并成功构建了p3×Flag-TRαA重组质粒。测序结果显示CDS区第850个核苷酸稳定由A突变为G, 导致氨基酸由Ser转变为Gly; 920—1030 bp有两小段, 分别是连续9 bp、连续11 bp与NCBI上传序列始终不同, 且不同的高保真酶扩增测序结果相同。
2.2 牙鲆TRαA基因推导氨基酸序列分析及蛋白结构预测
利用ProtParam tool (https://web.expasy.org/protparam/)平台, 分析TRαA基因编码序列所推导蛋白质的基本理化性质, 预测蛋白质的相对分子量为47 kD, 理论等电点是7.06。Leu、Lys、Glu、Ser含量比较高, 分别为 8.4%、8.2%、7.9%、7.5%。利用SWISS-MODEL (https://www.swissmodel.expasy.org/)在线软件, 分析TRαA的三级结构(图 1), 结果显示该蛋白含有8个不同长度的α 螺旋结构、1个典型的β转角和无规卷曲。
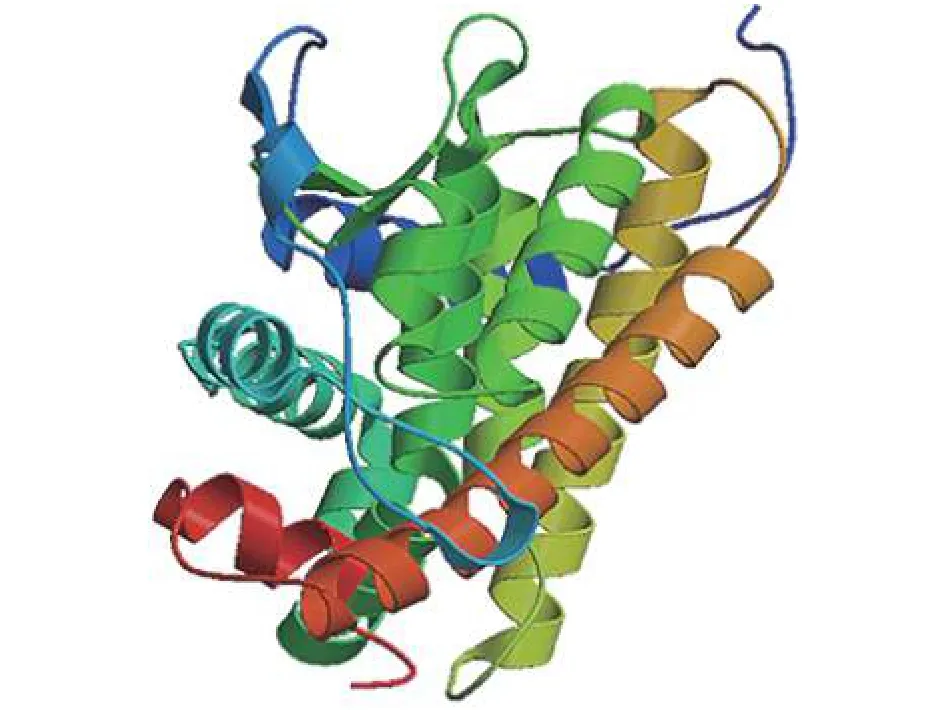
图 1 牙鲆TRαA受体蛋白结构预测Fig. 1 Prediction of protein structure of P. olivaceus TRαA receptor
2.3 TRαA基因的RT-PCR及实时定量PCR检测
RT-PCR产物电泳结果显示, 实验组在长度1200 bp处显示有一特异性条带, 与预期大小相同,而对照组均出现一个相应的亮度减弱且二者亮度相同的条带, 原因可能是HEK293T细胞中存在牙鲆TRαA基因的同源序列; 实时定量PCR也显示, 与对照组相比, 实验组TRαAmRNA表达水平显著升高,这都表明转染p3×Flag-TRαA的HEK293T细胞中, 成功转录出牙鲆TRαAmRNA(图 2)。
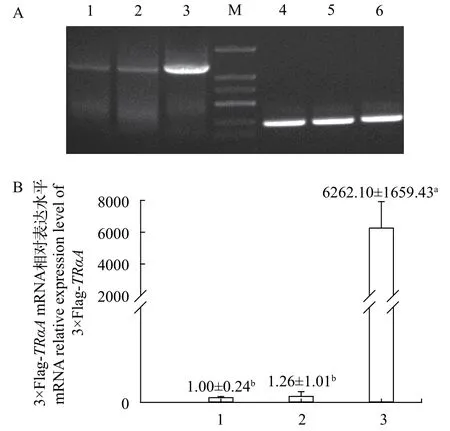
图 2 重组质粒或空载转染HEK293T细胞TRαA mRNA RTPCR及实时定量RT-PCR表达分析Fig. 2 RT-PCR and real time quantitative RT-PCR analysis of P.olivaceus TRαA mRNA in the HEK293T transfected with recombinant or vacant vector using 18S as an internal reference
2.4 融合蛋白3×Flag-TRαA的Western blot检测
BCA法(图 3)测得未转染、空载转染和重组质粒转染HEK293T细胞提取的总蛋白浓度分别是2.055、1.650和3.440 μg/μL, SDS-PAGE上样10 μg分离蛋白条带后, Western blot分析显示重组质粒转染组检测到一条50 kD大小条带, 其蛋白分子量与3×Flag-TRαA融合蛋白的大小相符; 而未转染及空载转染组未检测到目的条带(图 4)。原则上空载转染组可被抗Flag标签单抗特异性识别, 只因3×Flag蛋白分子与目标融合蛋白相比太小仅2.9 kD,分离胶底部被切除或已跑脱导致无法检测到。
2.5 目的蛋白的分离纯化
通过Western blot在洗脱液中可检测出50 kD左右的蛋白, 即通过亲和层析、过滤除菌得到了纯化的融合蛋白3×Flag-TrαA(图 5)。
2.6 候选靶启动子的筛选与重组报告载体构建
根据atoh8基因启动子转录因子结合位点预测结果, 分别设计了3个上游引物, 1个下游引物, 试以牙鲆基因组DNA 为模板扩增atoh8基因5′-侧翼序列的各缺失片段, 电泳结果显示其大小约为1500、1300和700 bp, 实际扩增出的片段大小为1517、1333和708 bp (图 6A为其实际扩增测序的序列, 引物与预测TREs位点已标出), 与NCBI上传的大小稍有出入, 并成功构建了重组质粒pGL3-Pro-atoh8-1517/1333/708。

图 3 BCA法测定蛋白浓度标准曲线Fig. 3 Determination of protein concentration standard curve by BCA method
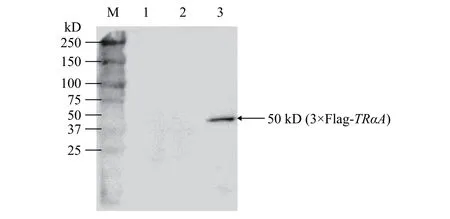
图 4 融合蛋白3×Flag-TRαA的Western blot分析Fig. 4 Western blot analysis of fusion protein 3×Flag-TrαA

图 5 纯化融合蛋白3×Flag-TRαA的Western blot分析Fig. 5 Western blot analysis of purified fusion protein 3×Flag-TRαA
2.7 atoh8基因启动子研究
根据预测TRE位点所在位置, 分别截取了3个缺失片段, 大小分别为1517、1333和708 bp, 依次包含3个、2个、1个TRE位点, 测序结果如图 6A所示,各缺失片段起止区段如图 6B所示。分别检测每一缺失片段的启动子转录活性, 对每一缺失片段进行3种处理(不加TRαA和T3、只加TRαA或同时加TRαA与T3)后再分别检测其对启动子转录活性的影响, 结果如下: 图 7A显示, 与pGL3-Basic转染组(阴性对照)相比, pGL3-Pro-atoh8-1517/1333/708转染组报告基因均表达, 表明Pro-atoh8-1517/1333/708都具有启动子转录活性, 且Pro-atoh8-1333活性最高, Pro-atoh8-1517活性最低; 图 7D显示, 与仅转染pGL3-Pro-atoh8-708相比, 同时转染pGL3-Proatoh8-708和p3×Flag-TRαA的报告基因的表达量无显著变化, 再加T3后也无显著变化, 说明此缺失片段不存在TRαA受体介导T3调控的TRE位点; 图 7C则表明, 与只转染pGL3-Pro-atoh8-1313相比, 同时转染pGL3-Pro-atoh8-1313和p3×Flag-TRαA的报告基因的表达量显著降低, 且这种降低在加T3后其又显著上调, 说明在Pro-atoh8-1333中存在1个TRE位点; 图 7B显示, 与只转染pGL3-Pro-atoh8-1517相比,同时转染pGL3-Pro-atoh8-1517和p3×Flag-TRαA的报告基因的表达量略有升高, 再加T3后其显著升高, 表明Pro-atoh8-1517存在2个TRE位点(包括Proatoh8-1313的1个TRE位点); 显然, 无论是Pro-atoh8-1517还是Pro-atoh8-1333, T3加入后都会诱导报告基因的转录水平显著升高, 即启动子转录活性显著增强, 因此说明, T3通过结合在atoh8基因启动子区(-1497— -688)特异的2个TRE序列上的TRαA受体来调控该基因的转录。
3 讨论
为了鉴定出TH-TRαA信号通路上直接调控的靶基因, 综合预测出候选靶基因atoh8(Gene ID:109627718), 克隆其5′-侧翼序列各缺失片段并成功构建pGL3-Pro-atoh8-1517/1333/708(分别含TRE位点: 3、2、1)。本研究克隆了牙鲆TRαA基因完整CDS区, 全长1251 bp (5′端多加1 bp保证TRαA受体正确翻译), 且TRαA受体DNA结合结构域(DBD)非常保守, 为调控下游靶基因的转录激活与抑制所必需。已有研究[7,18]证实, DBD功能域最保守, 内有2个锌指结构, 第一锌指内含有P盒, 为识别并结合TREs之AGGTCA(TRs半位)所必需; 第二锌指内含有D盒, 识别DR4元件(两半位之间间隔4个核苷酸)两半位(half-site)的间隔并参与受体二聚体的形成。RT-PCR、实时定量PCR和Western blot分析均表明成功构建p3×Flag-TRαA重组真核表达载体, 在目的蛋白N端融合Flag纯化标签, 由于3×Flag标签分子量相对较小, 因此几乎不会影响蛋白质的活性,纯化产物可直接用于蛋白功能的研究。3×Flag-TRαA的外源性真核表达特异性强, 具有翻译后加工修饰功能, 使其结构、糖基化方式更接近天然蛋白且生物活性稳定[19]。

图 6 报告基因质粒pGL3-Pro-atoh8-1517/1333/708示意图Fig. 6 Schematic diagram of the reporter gene plasmid pGL3-Pro-atoh8-1517/1333/708
进而为了验证TRαA受体与atoh8基因5′-侧翼序列是否存在相互作用, 针对上述截取的atoh8基因5′-侧翼序列的3个缺失片段(大小分别为1517、1333和708 bp, 依次包含3个、2个、1个TRE位点),及相应成功构建的重组质粒pGL3-Pro-atoh8-1517/1333/708, 采用双荧光素酶报告实验, 首先检测了atoh8基因3个不同长度(1517、1333和708 bp)的5′-侧翼序列对其启动子转录活性的影响, 其次针对每一缺失片段, 进行了3种处理: 不作任何处理(TRαA和T3都不加)、只加TRαA或同时加TRαA与T3, 分别检测其对启动子转录活性的影响。需要指出的是, 截取的atoh8基因5′-侧翼序列长度为708 bp时, 相关转染组结果显示TRαA受体和T3对其启动子活性并无显著作用, 故不进行分析讨论, 以下仅讨论关于pGL3-Pro-atoh8-1517/1333转染组的情况。研究表明, 与对照组相比, 上述2个缺失片段都具有启动子转录活性, 其中截取atoh8基因5′-侧翼序列长度为1333 bp时的活性最高, 是长度为1517 bp时的大约3倍。据报道, 并非是截取基因的5′-侧翼序列越长其启动子转录活性越高[20], 本文结果亦如此。与仅转染pGL3-Pro-atoh8-1313的实验组相比,同时转染pGL3-Pro-atoh8-1313和p3×Flag-TRαA的报告基因的表达量显著降低, 但这种降低可被T3显著上调, 表明此缺失片段存在TRαA介导T3调控的TRE位点。对于pGL3-Pro-atoh8-1517转染组, 虽然pGL3-Pro-atoh8-1517包含pGL3-Pro-atoh8-1333的TRE位点, 但在TRαA受体加入后, 与不作任何处理组相比, 其启动子转录活性反而略有升高, T3加入后又使得其启动子转录活性显著升高, 这提示在加入与pGL3-Pro-atoh8-1333转染组等量的TRαA受体后, pGL3-Pro-atoh8-1517 5′端第1个TRE位点与第2个TRE位点结合TRαA受体, 综合效应是第1个TRE位点中和了第2个TRE位点TRαA受体对靶基因转录水平的降低, 说明第1个TRE位点起了一个正向调节作用。也就是说, pGL3-Pro-atoh8-1517包含的5′端起第1个TRE位点与第2个TRE位点通过TRαA受体介导T3调控着atoh8基因的表达水平, 避免表达过强或不足, 这可能是机体实现精准调控atoh8基因表达的一种方式。但无论是pGL3-Proatoh8-1517转染组还是pGL3-Pro-atoh8-1333转染组, T3对atoh8的转录水平都是一种正向调节作用。

图 7 牙鲆atoh8基因启动子活性测定Fig. 7 Determination of promoter activity of P. olivaceus atoh8 gene
综上所述, 本研究结果表明pGL3-Pro-atoh8-1517包含2个TRE位点, 且TRαA受体通过DNA结合结构域(DBD)结合在atoh8基因5′调控区-1497—-688特异的TRE序列来调节该基因的转录, 由此证明atoh8是TRαA介导TH调控的直接靶基因。本研究为深入探究甲状腺激素受体TRαA介导甲状腺激素调控的信号通路提供了基础依据。
猜你喜欢
北方农业学报(2022年3期)2022-08-08
上海金属(2021年6期)2021-12-02
中国渔业质量与标准(2021年5期)2021-11-20
昆明医科大学学报(2021年3期)2021-07-22
江西农业学报(2021年4期)2021-04-20
食品科学(2020年4期)2020-03-11
河北渔业(2019年11期)2019-12-11
生物学通报(2019年3期)2019-02-17
电脑知识与技术(2018年19期)2018-11-01
西南医科大学学报(2015年1期)2015-08-22
