
儿童原发性1 型高草酸尿症临床及AGXT 基因突变分析
2020-04-04 10:50张佳仪蔡晓懿邓会英许自川李颖杰
临床儿科杂志 2020年3期
张佳仪 蔡晓懿 陈 椰 邓会英 许自川 李颖杰
广州市妇女儿童医疗中心肾内科(广东广州 510000)
高草酸尿症(primary hyperoxaluria,PH)是一种罕见的常染色体隐性遗传病,是因体内酶缺陷导致的乙醛酸盐代谢障碍,血、尿中草酸异常增高的遗传代谢性疾病,根据酶缺陷的不同,主要分为PH1、PH2和PH 3 三型,PH 1 是发病率最高的一种亚型,约占原发性高草酸尿症病例的80%[1-2]。PH1为罕见病,在我国的发病率未见明确报道。其临床无明显特异性,主要表现为酸碱失衡、电解质紊乱、多发性肾结石以及肾衰竭等,在婴儿期起病患儿往往涉及多系统损害,预后差。随着基因诊断技术的不断发展,临床上对PH认识的逐渐提高,目前发现其主要致病基因为AGXT。本研究通过总结5例PH1患儿的临床特点,并对其基因结果进行分析,旨在进一步提高对儿童PH1的认识。
1 临床资料
2016 年10月至2019 年3月在广州市妇女儿童医疗中心肾内科诊断为PH 1 患儿5 例。其中男性3 例、女性2例,发病年龄2个月至4岁,诊断年龄2个月至9岁。5例患儿就诊时均有不同的临床表现。2例患儿以面色苍白就诊,3 例患儿以尿少、浮肿就诊。2 例患儿(例1和例2)以急性肾衰竭起病,并累及多器官损害,透析治疗效果差,在起病后3个月内死亡。3例患儿(例3、例4和例5)具有不同临床表现的慢性病程,感染后出现急性肾衰竭的临床表现;其中例5 患儿在4 岁体检时影像学发现肾结石,当时无肾功能损害及相关临床表现,9岁时因感染后出现乏力、少尿就诊,尿肌酐提示已进入尿毒症期。例5 患儿有肾结石家族史,其余患儿均无相关家族史。
5 例患儿实验室检查均有不同程度的电解质紊乱、代谢性酸中毒及蛋白尿。5例患儿肾脏B超均有异常回声,1例患儿腹部X片有多发性结石灶(图1)。例4患儿肾穿刺活检提示高草酸尿症。见表1、2。
PH 1 的诊断的金标准是肾活检标本中AGT 酶的活性测定,但是由于肾穿刺活检是一项有创检查,患儿及其家属的接受度较低,随着生物分子学及基因技术的发展,目前基因检测技术已成为PH 1 诊断的首要方法。经医院医学伦理审查,家属知情同意后,采集5 例患儿及其父母外周血标本,采用盐析法提取基因组DNA,通过检索数据库及相关文献设计肾病相关基因二代测序,筛选与受检者临床表型相关的高度可疑变异基因,通过Sanger测序法对患儿及父母进行验证。5 例患儿均发现AGXT基因突变。例1 检测到1个纯合突变,为11号外显子c.1079G>A突变,导致第360 号氨基酸由精氨酸变异为谷氨酰胺(p.R 360 Q),为错义突变,其父、母、兄该位点均为杂合变异(图2)。例2 检测到2 个杂合突变,分别为1 号外显子c.2 T>C突变,导致第1 号氨基酸由甲硫氨酸变异为苏氨酸(p.M 1 T),为错义突变,其父该位点杂合变异;8 号外显子c.815_816 insGA(插入鸟嘌呤腺嘌呤),导致氨基酸改变(p.S275Rfs*38),为移码突变,其母该位点杂合变异(图3)。例3 检测到2 个杂合突变,分别为2号外显子c.215A>T,导致第215号核苷酸由腺嘌呤变异为胸腺嘧啶,导致天冬酰胺变异为异亮氨酸(p.N72I),为错义突变,其父该位点杂合变异;6号外显子c.679_680+2delAAGT,导致氨基酸改变剪接体,为移码突变,其母该位点杂合变异(图4)。例4检测到2个杂合突变,分别为6号外显子c.679_680del缺失,导致氨基酸移码突变(p.K228Efs*26),其父该位点杂合变异,其母、兄该位点无变异;2号外显子c.190A>T突变,导致第64号氨基酸由异亮氨酸变异为苯丙氨酸(p.I64F),为错义突变,其母、兄该位点杂合变异,其父该位点无变异(图5)。例5检测到1个纯合突变,为6号外显子c.679_680delAA(缺失),导致氨基酸改变(p.K 228 Efs*26),为移码突变,其父、母该位点杂合变异(图6)。
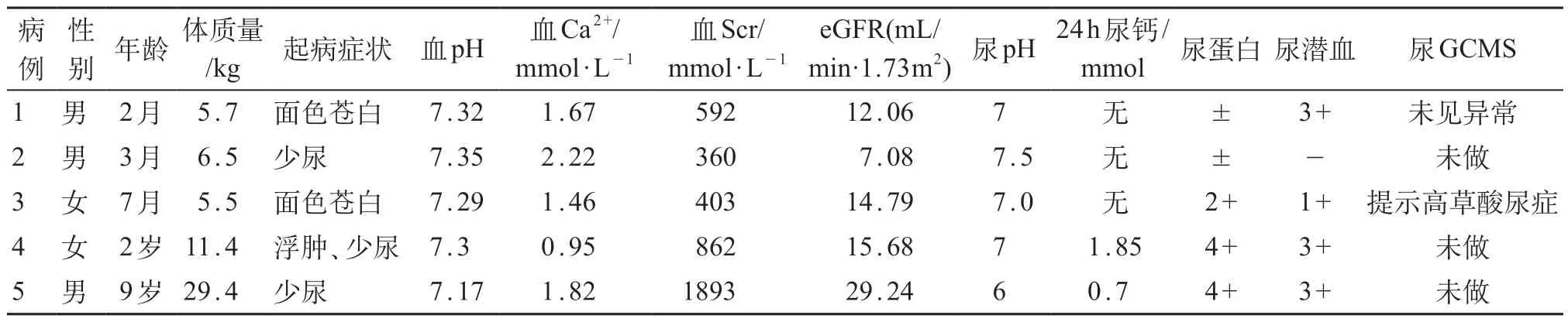
表1 PH1患儿主要临床资料和实验室检查

表2 PH1患儿影像学检查结果
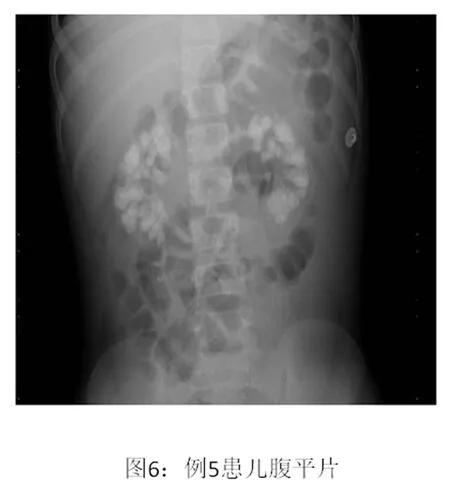
图1 例5 患儿腹平片
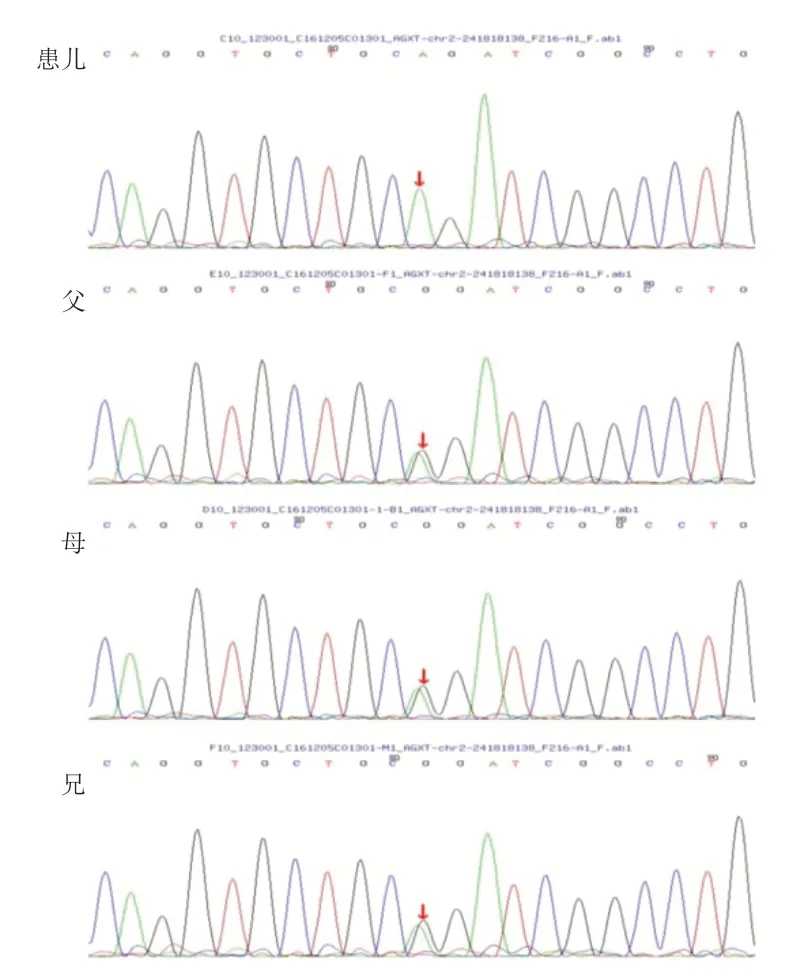
图2 例1 家系AGXT 基因外显子11 部分序列
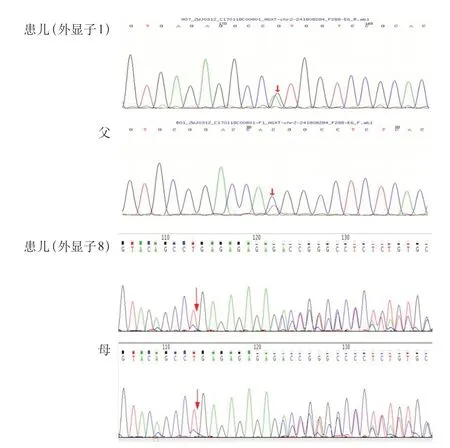
图3 例2 家系AGXT 基因外显子1、8 部分序列
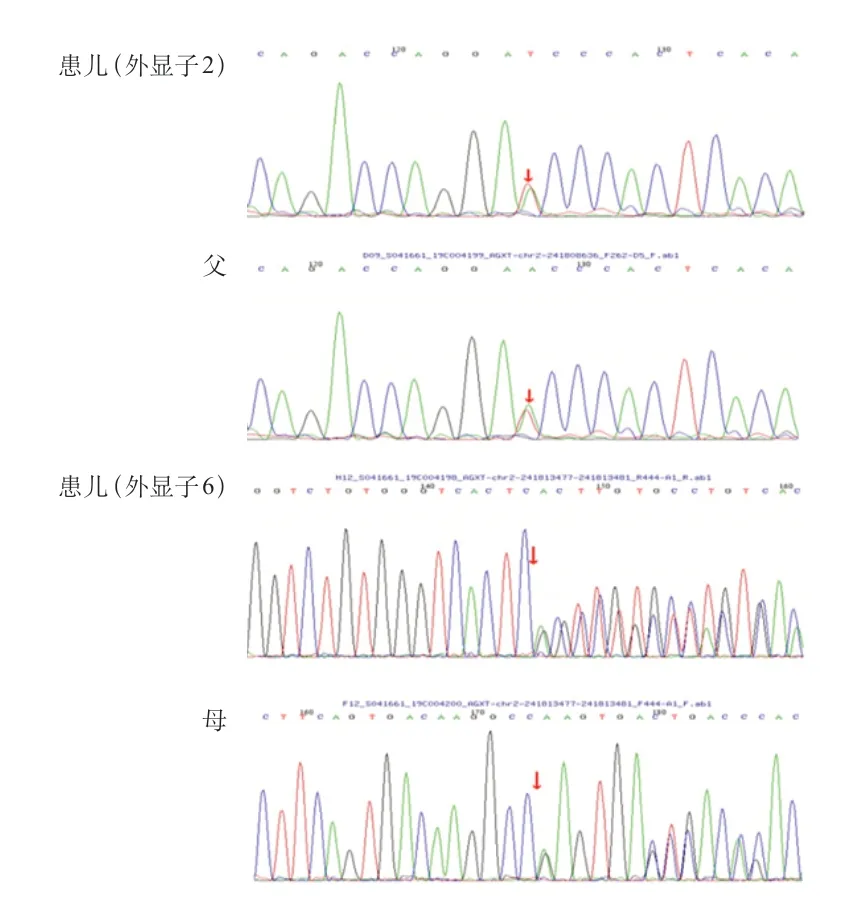
图4 例3 家系AGXT 基因外显子2、6 部分序列
2 讨论
PH1几乎可发生于任何年龄,从出生到60岁均可发生,据文献报道其平均发病年龄为(9.5±10.2)岁(中位年龄5.5 岁)[2]。基于人口统计学研究,PH 1 在欧洲及北美人群的患病率为1/106~3/106[2];在中欧地区,每12 万个新生婴儿中大约有1 例为PH 1[3];在美国、日本PH 1 占儿科终末期肾脏病的1%~2%[4]。PH1的症状主要与肾脏中草酸盐沉积有关,包括尿石症和肾钙质沉着症,并伴有进行性肾衰竭。由于草酸盐不断沉积,导致肾功能逐渐下降,肾脏不能排出草酸盐,使血浆中的草酸钙水平上升,沉积在各个组织,表现出全身性高草酸病。
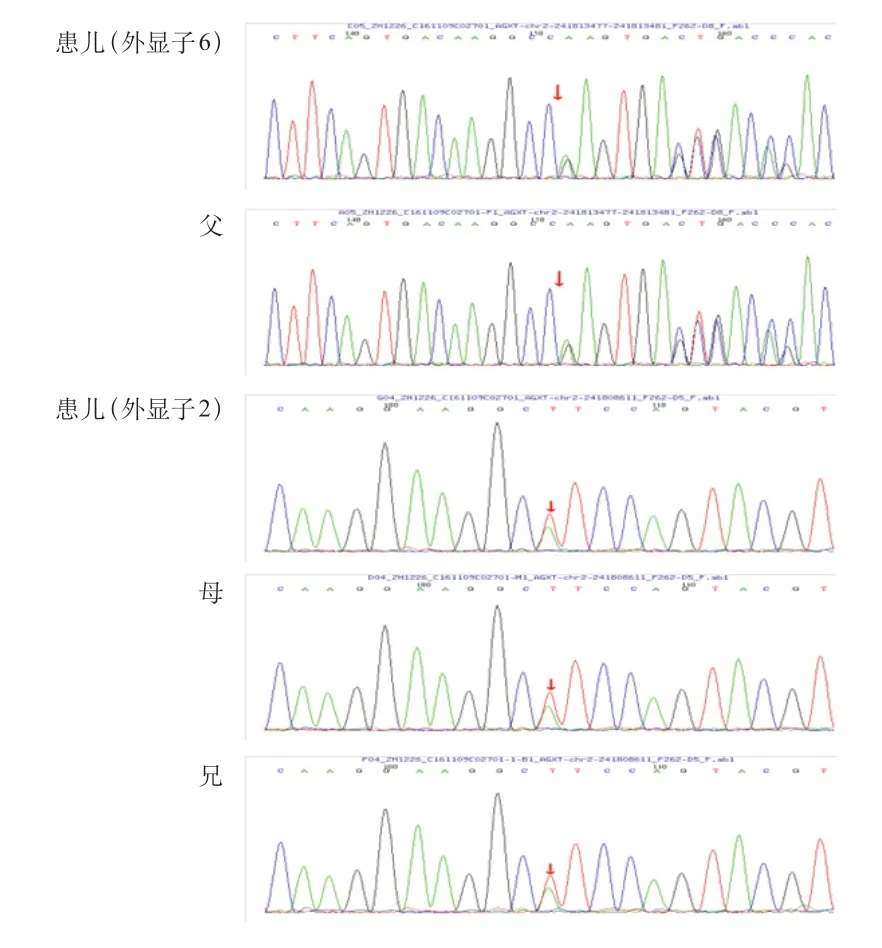
图5 例4 家系AGXT 基因外显子6、2 部分序列
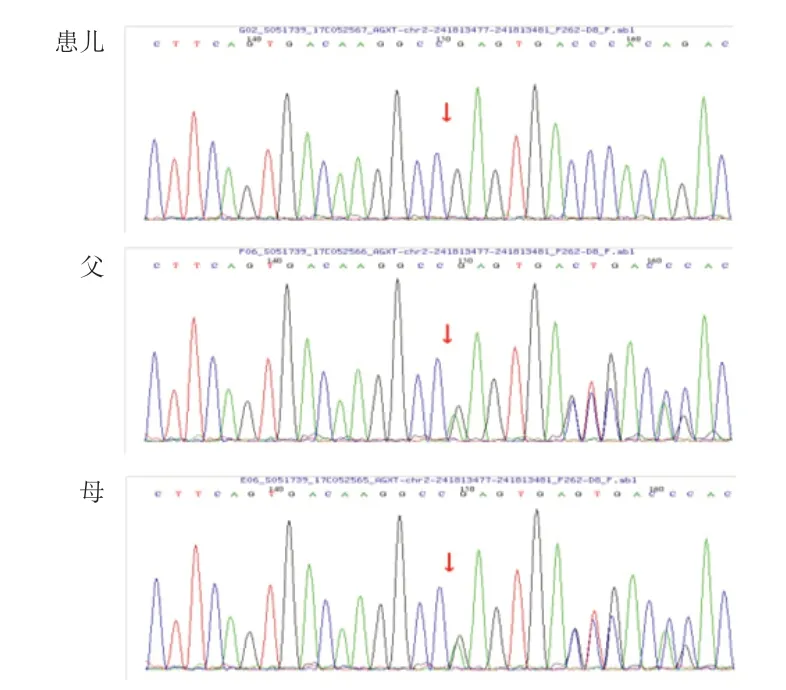
图6 例5 家系AGXT 基因外显子6 部分序列
PH1临床表现可以多种多样,部分患儿早期可无临床症状,也可以呕吐、腹泻、血尿、贫血就诊,部分患儿甚至就诊时已表现为终末肾病期(end stage renal disease,ESRD)[5-6]。成年期起病的PH1的ESRD者通常只有散发性肾结石的病史,但超过50%患者在诊断时或诊断前已经进入ESRD[7-9]。在婴儿期发病的PH1主要呈现的特征是代谢性酸中毒,以及急性肾衰竭,约10%的患儿在生后6个月内可出现症状,同时伴有严重的视网膜病变等其他多器官损害[10]。本组5例患儿中有3 例在婴儿期起病,其中2 例起病初即表现为急性肾衰竭,伴有顽固性代谢性酸中毒、高钾血症、低钙血症及多器官损害,肾脏B 超提示病理性改变,但是还未出现肾结石的病理表现,可能与患儿年龄小有一定相关性。这类患儿病情进展快,在起病后3 个月内死亡[11]。本组例5 患儿在学龄前起病,早期无明显肾功能损害,随着肾钙不断沉积表现为多发性肾结石,出现梗阻性肾病的表现,肾损害急剧进展,就诊时已进入ESRD,现予透析治疗维持中。这例患儿有肾结石家族史,其姐姐亦存在有多发性肾结石,但是无相关临床表现,肾功能无异常。由于患儿姐姐没有进一步行基因检测,两者临床表现异常的原因推测可能有与基因突变位点不同有关,具体机制有待进一步研究分析。
PH1 是由于肝脏特异性过氧化物酶丙氨酸乙醛酸氨基转移酶(alanine-glyoxylate aminotransferase,AGT)的先天性缺陷所致,AGT 酶的编码基因为AGXT基因,是目前所知的与PH1发病相关的基因[12]。人类AGXTcDNA序列在1990年被首次克隆[13]。目前已发现200多个AGXT突变位点(http://www.uclh.nhs.uk/phmd),其中包括单个核苷酸的替换(错义突变、无义突变及同义突变)、移码突变、剪接子突变、大的插入或缺失、小的插入或缺失等[10]。AGXT基因不同的突变位点通过不同的分子机制引起不同的表型,例如p.G170R和p.F152I变体是造成过氧化物酶体错误靶向运输[14-15]。
p.F152I、p.G170R、p.I244T和c.33_34insC是PH1的常见突变,占PH1等位基因的50%以上[16-18]。本组5例患儿均未携带这些常见变异,但检测到AGXT基因的6个突变,包括1个新的突变c.190A>T(p.I64F)。3例患儿检测出c.679_680del突变,分别是c.679_680+del和c.215 A>T(p.N 72 I)、c.190 A>T(p.I 64 F)复合杂合突变,c.679_680+del纯合突变;因此,在本组患儿中,c.679_680+del突变占4/10 等位基因,之前已有c.679_680+del 纯合突变在中国人群中的报道,故有提出将c.679_680+del突变作为中国人群中的常见突变之一[19-20]。c.679_680 del 位于6 号外显子,研究表明,6 号外显子的编码序列相对保守,对AGT 酶发挥催化活性起着关键作用,其核苷酸缺失会引起移码突变,导致mRNA剪切错误引起氨基酸残基位置改变[16]。此外,本组患儿中,例2为c.815_816insGA和c.T>C(p.M1T)杂合突变。根据先前报道,c.815_816insGA突变可能是中国PH1患者的热点突变之一。目前已报道c.815_816insGA(p.S275Rfs * 38)和c.33_34insC(p.K12Qfs * 156)[19],c.242C> A(p.S81X)[21],c.346G>A(p.G116R)[22],c.364C> T(p.R122X)[23],c.1015delG(p.V 339 Sfs*2),c.473 C>T (p.S 158 L),c.614 C>T(p.S205L)[24]复合杂合突变。但基因型和表型的关系仍不清楚,携带相同变异位点,如c.815_816insGA的不同患者表现出不同的临床特点和预后。
PH1早期干预的目的主要是缓解肾功能损害,一旦诊断考虑PH1就应该及时给予支持治疗,减少草酸合成减轻肾脏负担,主要包括水化碱化、维生素B 6等治疗;限制草酸盐摄入对于减少草酸形成的作用是有限的,因为PH患儿草酸盐的主要来源是内源的,肠道草酸盐的吸收量很低[25]。对于有阻塞、感染或多发性尿结石为主要临床症状的患儿可考虑外科干预以缓解症状。PH1患儿一旦进入ESRD后则需要依靠肾脏替代治疗以等待移植时机,目前治疗PH 1 最有效的手段是肝肾联合移植,既能解决AGT缺乏,又能减少肝脏草酸合成,同时又能使组织中沉积的草酸钙溶解、析出,经移植肾排泄,进而逆转系统性草酸盐沉积症状[26-27]。
综上所述,本组5 例患儿的临床表现以肾脏损害为主,并伴有顽固性代谢性酸中毒、高钾血症、低钙血症及多器官损害;基因检测发现2个中国人群常见的突变c.815_816insGA、c.679_680del,以及1个新的突变c.190A>T。临床疑似PH1患儿应尽早完善基因检查,及早干预,争取延缓病情进展。
猜你喜欢
昆明医科大学学报(2022年7期)2022-11-24
化工生产与技术(2022年3期)2022-07-01
种子(2021年3期)2021-04-12
肾脏病与透析肾移植杂志(2021年1期)2021-01-13
中小企业管理与科技(2019年18期)2019-08-06
中国医药指南(2018年30期)2018-11-16
中国生育健康杂志(2018年6期)2018-11-13
科技视界(2016年27期)2017-03-14
饮食科学(2016年3期)2016-07-04
饮食科学(2016年3期)2016-07-04
