
丙酮酸激酶缺乏症2 家系基因变异分析
2020-03-12 04:04:08程首超
临床儿科杂志 2020年2期
邱 局 程首超
咸宁市中心医院 湖北科技学院附属第一医院(湖北咸宁 437100)
丙酮酸激酶缺乏症(pyruvate kinase deficiency,PKD)是一种常见的遗传性红细胞酶相关疾病,可引起遗传性非球形红细胞溶血性贫血,是仅次于葡萄糖-6-磷酸脱氢酶缺乏症的最常见红细胞酶病,为常染色体隐性遗传[1-2]。丙酮酸激酶是糖酵解过程中关键的限速酶之一,催化磷酸烯醇丙酮酸转变为丙酮酸同时产生ATP 供能[3]。PKLR基因编码丙酮酸激酶关键同工酶,该基因异常可导致红细胞对钾离子通透性增强,致使红细胞膜钠钾泵供能障碍,最终导致红细胞在肝脾及骨髓内破坏发生溶血。PKD临床诊断较为困难,基因检测可协助临床诊断。本研究通过对2 个PKD家系的回顾分析,探讨PKD的临床及致病基因突变特点。
1 临床资料
例1,女,3 岁8 个月,因面色苍白3 年余就诊。患儿生后有新生儿高胆红素血症,予以蓝光照射治疗。5 个月时因皮肤、巩膜黄染加重在外院输血治疗后好转。半年前因肺炎入院,发现血红蛋白(Hb)<50 g/L,再次输血治疗。患儿父母身体健康,无类似疾病家族史。体格检查:面色及巩膜黄染,口唇苍白,身高92 cm(-2SD),体质量11.5 kg(-2SD),多动,语言发育落后于同龄儿童,腹部平坦,肝脾肋下未触及。实验室检查:血常规红细胞2.34×1012/L,Hb 60 g/L,平均红细胞体积(MCV)92.0 f L,网织红细胞(RET)0.086,血清铁 32.5 μmol/L,铁蛋白 136.5 μg/L,Coombs实验阴性,Hams 实验阴性;血涂片示红细胞大小不均、偏大,可见嗜多色性红细胞。
例2,女,3岁10个月,因面色苍白,反复咳嗽2周就诊。患儿6月龄体检时提示贫血,1岁6个月时因肺炎入院发现Hb<60 g/L,输血治疗后贫血好转。住院期间骨髓细胞学检查提示增生性贫血。患儿父母体检未见异常,无类似疾病家族史。患儿平时体弱。入院体格检查:全身皮肤发黄,口唇、甲床苍白,巩膜黄染;身高93 cm(-2SD),体质量12.2 kg(-2SD),语言、运动发育可;肝肋下3 cm,脾肋下3 cm。实验室检查:红细胞2.87×1012/L,Hb 56 g/L,MCV 90.0fL,RET 0.057,血清铁33.8 μmol/L,铁蛋白 225.5 μg/L,Coombs实验阴性;血涂片示红细胞轻度大小不均,余未见明显异常。腹部B超提示胆囊结石。
为进一步明确诊断,经咸宁市中心医院医学伦理学委员会审核批准,家属知情同意,对患儿进行致病基因检测。采集患者及其家属的外周静脉血各2 mL,委托第三方公司进行全外显子(WES)测序,并利用Sanger测序验证。以全血基因组核酸提取试剂盒提取基因组DNA,致病基因PKLR的引物由上海生工合成;PCR反应体系扩增产物进行Sanger测序分析。结果发现,例1患儿PKLR基因存在复合杂合突变,c.106G>T以及c.817C>T(NM_000298),其中c.106G>T突变遗传自父亲,导致基因编码的蛋白36位由甘氨酸突变为丝氨酸(p.Ala 36 Ser);c.817 C>T 错义突变遗传自母亲,导致编码273 位氨基酸由精氨酸突变为半胱氨酸(p.Arg273Cys);患儿的弟弟及姑姑均为c.106G>T突变携带者。见图1。例2患儿PKLR基因存在复合杂合突变,c.1279G>T以及IVS6-1G>T,其中c.1279G>T突变遗传自母亲,使编码的427 位氨基酸由谷氨酸突变为终止密码子(p.Glu427Term);IVS6-1G>T剪接位点突变遗传自父亲;患儿的姑姑为IVS 6-1G>T突变携带者。见图2。检索数据库及文献发现,c.106G>T和 c.1279 G>T 为已知的致病突变(HGMD 数据库中有收录);IVS 6-1 G>T 突变位于经典的保守剪接位点区域,依据ACMG指南,为致病性证据非常强的突变,突变可引起PKLR基因RNA异常剪接;c.817C>T突变在千人基因组以及HGMD 数据库中未收录,mutationtaster在线软件预测为致病性改变,Polyphen2及SIFT软件预测突变影响蛋白质的功能。
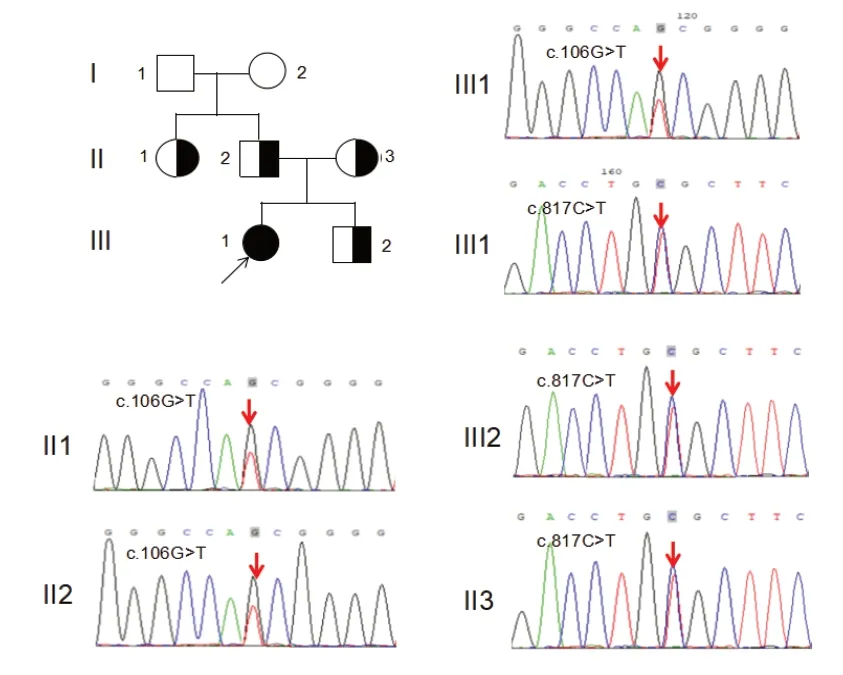
图1 例1 家系图及PKLR 基因测序图
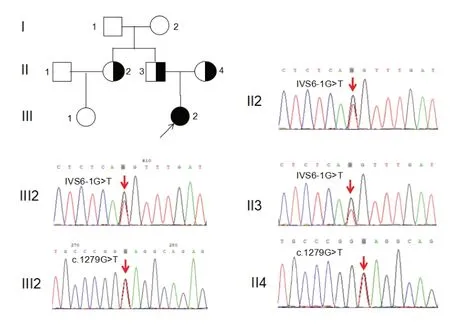
图2 例2 家系图及PKLR 基因测序图
取患儿新鲜静脉血1 mL,裂解红细胞后得到白细胞,利用Trizol 方法提取总RNA,经过反转录得到cDNA,利用PKLR基因特异性生物引物进行相对定量分析(以GAPDH 为内参基因)。结果显示,相对于同龄健康对照,例1 的PKLR基因表达量未见异常,例2的PKLR基因表达水平显著下降。推测例1的2个错义突变未影响PKLR基因转录,而例2 的剪接位点突变可能影响基因表达,荧光定量PCR结果显示与同龄的正常对照相比,例2 的PKLR基因mRNA 水平显著降低(P<0.001)
2 讨论
PKD的贫血严重程度不一,部分患儿在新生儿期即发生高胆红素血症,需要输血治疗。随着年龄增长,PKD 的贫血程度可减轻[4]。成年PKD 患者贫血症状较为稳定,但在感染、疲劳等条件下贫血可加重。此外PKD患者也常伴有黄疸、脾大以及胆囊结石等症状[5]。红细胞酶活性测定是诊断PKD的金标准,通过酶活性测定可帮助大多数患者明确诊断,但仍有部分患者酶活性变化不显著而需要基因检测或其他方法协助诊断[6]。本研究由于实验室条件限制,未检测红细胞酶活性。
PKD在全球均见报道,但多数分布在北欧地区[7]。PKLR基因突变是导致PKD的常见病因。截止到目前,已经报道的PKLR基因约有300种,包括错义突变、无义突变、碱基插入缺失等类型,且存在热点突变。美国、欧洲地区1529A和1456T突变常见,而亚洲地区1468T为热点[6,8]。
本组2 例患儿均表现为面色苍黄,皮肤黄染,贫血貌,均有输血史。实验室检测发现Hb严重低下,骨髓涂片显示增生性贫血。对患儿及其父母等进行基因检测,发现2 例患儿的PKLR基因均存在复合杂合变异。例1为c.106G>T以及c.817C>T 复合杂合突变,其中c.106G>T是已经报道的致病突变,而c.817C>T文献及数据库未见收录。生物信息学分析c.817 C>T在不同的物种之间保守、且突变影响蛋白的功能,依据ACMG指南,其可能为致病突变;例2为c.1279G>T和IVS 6-1 G>T 复合杂合突变,其中剪接位点突变IVS 6-1 G>T 位于经典的剪接区域(内含子6 的3’受体端),依据RNA加工成熟机制推测其可能导致外显子跳跃,影响mRNA 成熟。对患儿外周血PKLR基因表达分析发现,相对于同龄正常对照,例1的PKLR基因表达水平未见明显异常,而例2 的PKLR基因表达显著下降,推测剪接位点突变影响mRNA 成熟,导致RNA 易降解。依据患儿的临床表现以及基因检测结果,2例患儿均可诊断为PKD。
目前PKD的主要治疗方法为维持Hb水平稳定的输血治疗、脾切除治疗以及造血干细胞移植[9]。通过红细胞输注可使Hb水平维持在70~90 g/L的水平,但需要长期治疗以满足患儿的生长和发育;脾切除已被证实可有效提高PKD的Hb水平及网织红细胞总数[10]。理论上,造血干细胞移植可治愈PKD。研究发现,采用同胞供者骨髓干细胞移植治疗PKD,3年后患儿Hb水平及丙酮酸激酶活性均正常[11],但仍需要进一步的研究证实。本组2例患儿目前均接受红细胞输注治疗,需要进一步观察预后。
综上所述,PKD 临床表现为不同程度的贫血,皮肤、巩膜黄染等,疾病相关基因检测可帮助临床尽早诊断,并进行对症治疗及遗传咨询。此外,发现2个未见报道的PKLR基因突变,丰富了基因突变数据库,为今后该疾病的研究提供一定的参考。
猜你喜欢
工业微生物(2024年1期)2024-02-29 07:36:50
生物加工过程(2023年6期)2023-12-11 03:27:52
牡丹江医学院学报(2021年5期)2021-12-05 08:01:51
种子(2021年3期)2021-04-12 01:42:22
浙江中医杂志(2019年3期)2019-01-05 23:46:19
祝您健康·文摘版(2018年6期)2018-10-21 18:28:47
护士进修杂志(2017年2期)2017-02-16 06:40:18
蚌埠医学院学报(2016年7期)2016-09-01 06:44:13
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
