
唾液酸糖苷化的研究进展
2020-03-06 09:13陈鲜丽赖小将张福展
海峡科技与产业 2020年11期
钟 鸣 陈鲜丽 陈 艳 陈 领 赖小将 张福展
韶关学院医学院,广东 韶关 512026
唾液酸(SA),学名为“N-乙酰基神经氨酸”,是一种天然存在的碳水化合物。它最初由颌下腺粘蛋白中分离而出,也因此而得名。唾液酸通常以低聚糖,糖脂或者糖蛋白的形式存在。人体中,脑的唾液酸含量最高。脑灰质中的唾液酸含量是肝、肺等内脏器官的15 倍。
大量研究表明,唾液酸在抗炎、抗病毒和抗肿瘤等方面呈现出显著的生物学功效[1],尤其在抗流感方面。流感是一种急性呼吸道传染病,具有传染性强、传播速度快、流行面广、发病率高的特点。根据中国疾病预防控制中心数据显示,仅2019 年3 月份,全国共报告流行性感冒病例358757例,死亡31 例[2]。不仅如此,由流感引发的肺炎及心、肾等多种脏器衰竭更可以导致老人、儿童等高危人群死亡。近年来,随着新的高致病性禽流感和猪流感在全国及世界范围内的传播,各国政府均要求各国疾控中心(Center for Disease Control and Prevention,CDC)建立流感观测点,定期发布流感疫情情况报告,并加强了对流感的研究力度。我国是人口大国,庞大的人口基数使得对流感的监控与防治具有重大社会和国家安全意义。
流感由流感病毒引起,流感病毒属正粘病毒科(Orthonlyxovoridae)RNA 病毒,病毒颗粒直径80 ~120nm。根据RNA 基因的不同,人流感病毒分为甲(A)、乙(B)、丙(C)三种类型。流感病毒表面有两种重要的糖蛋白受体,血凝素(Hemagglutinin,HA)与神经氨酸酶(Neuraminidase,NA),二者是流感病毒亚型划分的主要依据,同时也是抗流感药物设计的主要靶点。临床上主要以接种流感疫苗和使用抗流感药物进行流感的预防与治疗[3]。由于针对HA 的流感疫苗的制备、储藏及运输的苛刻条件,同时由于抗原性漂移(Antigenic Drift)的存在,使得有效性与及时性存在诸多问题,因此抗流感药物成为防治流感爆发的主要手段。针对流感病毒表面神经氨酸水解酶(亦称唾液酸水解酶,Neuraminidase,NA)的一类抑制剂是迄今为止最有效、最成功的设计案例(见图1)。
流感病毒表面的糖蛋白受体血凝素能与多种动物(如鸡、豚鼠)和人的红细胞表面的末端唾液酸糖蛋白结合,介导病毒的融合,因此,利用唾液酸糖苷化反应模拟生物结合,对于抗流感病毒药品的研制有重要意义。随着人们对唾液酸在生物体内的生理功能逐步深入了解,开展唾液酸糖苷的化学合成与生物活性研究便有了非常重要的意义。与普通的糖苷化反应相比,唾液酸进行糖甘化反应所需的条件更为严格,并且其立体构型极难控制。
唾液酸C-2 卤化物是最早用于唾液酸糖苷化反应的物质之一,且在20 世纪六七十年代被广泛使用,不过目前仅被运用于一些简单的糖苷化反应实验中。为了提高对唾液酸糖苷化反应的研究效果,越来越多性质各异的离去基团被用于糖苷化反应之中。其中包括长链烷基唾液酸硫苷[4]、唾液酸β-亚磷酸酯[5]和三氟乙酰亚胺酯[6]等,相关研究发现,以上方式均可简单快捷地对相应唾液酸糖苷进行制备,并可明显地提高反应产率和立体选择性。

图1 唾液酸及基于唾液酸的各类药物的结构
有项目组研究表明,在使用N-Fmoc(9-芴基甲氧基羰基保护基)、N-Troc(2,2,2-三氯乙氧基羰基)及N-TFA(三氟乙酰基)对唾液酸C-5 位氨基进行保护的过程中发现,N-Fmoc 衍生物的产率较低(27%),其它相对较好,可以达到63%~89%的产率;与一级醇发生糖苷化反应,选用N-碘代丁二酰亚胺/三氟甲磺酸作为催化剂后,N-Fmoc、N-Troc 或N-TFA 保护的唾液酸衍生物糖苷化产率均比较理想(>90%)。由于脱除TFA 保护基需要较强的碱性条件,这将会破坏目标分子的构型,因此,选用N-Fmoc,N-Troc 保护基较TFA 更为合适,而N-Troc 保护的唾液酸又比N-Fmoc 保护操作更容易一些,因此N-Troc 作为保护基用于目标分子的全合成研究具有更多的选择优势[7-8]。
在流感爆发季节,抗流感药物成为了防治的主要手段。目前上市的作为抗流感一线药物的NA 抑制剂有两种:葛兰素公司的Zanamivir (商品名Relenza,中文商品名乐感清)与罗氏公司的Oseltamivir(商品名Tamiflu,中文商品名达菲)。另有强生公司和BioCryst 公司合作开发的Peramivir 已经进入III 期临床,并获得FDA 紧急使用授权;日本FDA 也已经批准Laninamivir 和Peramivir 进入III 期临床。仔细分析他们的结构不难发现,上述的上市药物均为唾液酸经过化学改造的结构类似物,其与NA 的亲和力相比于NA 的天然配体唾液酸更强。尽管此类药物在临床上的应用已经取得巨大成功,但是流感毒株耐药性问题却日益凸显。美国疾病控制与预防中心报告指出, 临床长期治疗的儿童体内经常发现对Zanamivir 敏感性降低的病毒株,另一方面,耐达菲的流感病毒数量也正在逐年增加,因此药物化学家仍然不懈地寻找基于唾液酸的结构新颖的NA 抑制剂。Kim 小组报道了双氟取代Zanamivir 类似物DFSAs,结果显示对耐药株有良好的抑制活性,相关研究正在进一步深入。本课题组希望在此基础上,对DFSAs 进行进一步的结构改造,以期获得更加优良的抗耐药效果,初步的设计是在DFSAs 的C-7 位引入叠氮基团,以炔基修饰的乙二醇、季戊四醇、乳糖为骨架,合成二价、四价、八价DFSAs缀合物。
由于NA 的主要功能是水解唾液酸糖苷键,阻止病毒聚集,协助子代病毒粒子从感染的细胞中释放,故制备正常的唾液酸氧苷显然会遭到NA 的破坏,无法达到预想的目的。因此,我们的另外一种思路是考虑采用杂原子(例如氮原子或者硫原子等)替换唾液酸糖苷键中的氧原子,以此来构建唾液酸伪糖苷。由于NA 在识别这种伪糖苷的过程中无法水解糖苷键,没有破坏糖环结构,因此唾液酸伪糖苷在起到蛋白识别作用的同时,能够对NA 的水解起到抵抗作用,化合物更加稳定。后面这种思路是我们项目组正在进行的工作。
通过一系列比较,我们认为,用氮原子替换唾液酸糖苷键中的氧原子,合成方面难度较小,并且有利于后续的连接臂合成,我们设计的氮原子替换唾液酸糖苷键氧原子形成的化合物结构如图2 所示。

图2 位氨基取代的唾液酸
接下来,就可以用各式各样的连接臂来衔接伪糖苷键,例如,选择酰胺连接来进行伪糖苷的构建,方便后续与数量庞大的生物或者人工合成的高分子进行组装,该分子的结构如图3 所示。
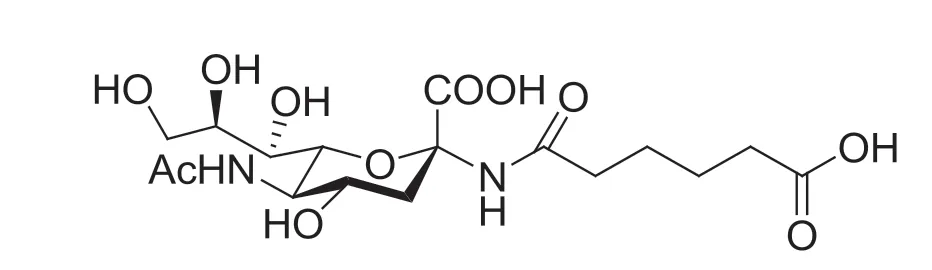
图3 酰胺连接的唾液酸伪糖苷
上述制备的唾液酸伪糖苷与牛血清白蛋白HSA 及聚醚酰亚胺PEI 结合,构建糖缀合物,用来模拟细胞表面的糖蛋白结构,进行糖及蛋白受体相互结合的作用研究。由于HSA 和PEI 具有方便易得、生物可降解、无毒、无免疫原性等特性,故我们选择它们作为骨架构建糖缀合物。构建的糖缀合物结构见图4。
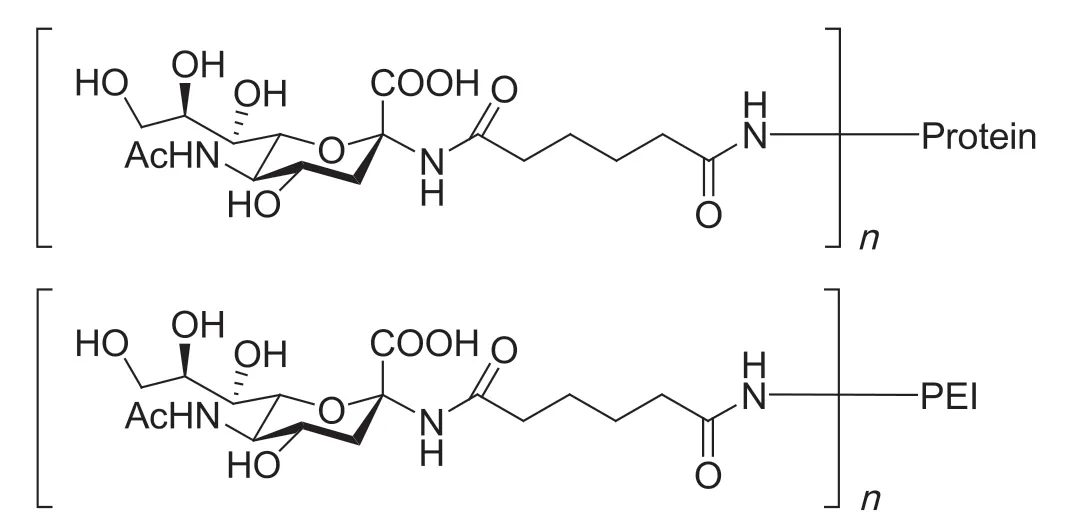
图4 HSA 和PEI 为骨架构建的糖缀合物

图5 唾液酸氨基甲酸酯、酰胺和硫脲连接半乳糖的合成
除此之外,我们将2 位氨基取代的唾液酸进行全乙酰化保护,进一步合成唾液酸氨基甲酸酯、酰胺和硫脲寡糖,对上述提到的三类化合物进行组装,其中重要的反应步骤如图5 所示:
通过对唾液酸糖苷化反应进行探讨,合成不同结构的唾液酸糖苷化给体,旨在提高唾液酸糖苷化反应的产率以及立体选择性。本项目组设计用杂原子替换唾液酸糖苷键中的氧原子,构建唾液酸伪糖苷,该合成的唾液酸伪糖苷是新化合物,暂未见文献报道,在起到蛋白识别作用的同时,能够对NA 的水解起到抵抗作用。通过总结糖苷化反应的进展,为我们项目组利用糖苷化反应来制备糖缀合物,增加其与流感病毒表面糖受体之间的结合,并以此提高抑制流感病毒活性,提供了坚实的实验参考价值。
猜你喜欢
现代食品科技(2023年12期)2024-01-09
食品工业科技(2019年5期)2019-04-01
中成药(2018年7期)2018-08-04
中成药(2018年1期)2018-02-02
现代检验医学杂志(2016年1期)2016-11-12
分析科学学报(2015年3期)2015-10-18
大连医科大学学报(2015年6期)2015-03-22
中国当代医药(2015年33期)2015-03-01
四川师范大学学报(自然科学版)(2014年4期)2014-10-09
食品科学(2013年17期)2013-03-11
