
淀粉样β蛋白的微生物表达及其在抑制剂筛选中的应用
2019-11-20 02:28刘夫锋赵文平路福平
微生物学杂志 2019年5期
刘夫锋, 赵文平, 位 薇, 王 英, 李 丽, 路福平
(1.天津科技大学 生物工程学院,天津 300457;2.天津科技大学 海洋与环境学院,天津 300457)
淀粉样蛋白的错误折叠和聚集会导致多种蛋白质构象疾病,例如帕金森综合症、II型糖尿病、疯牛病、亨廷顿病和阿尔茨海默病(Alzheimer′s disease,AD)等[1]。这类疾病的共同特征是细胞内或细胞外的蛋白质错误折叠和聚集所形成的淀粉样斑块。其中,AD是目前世界上影响范围最广的蛋白质构象疾病。据2019世界阿尔茨海默病组织统计,2018年全球AD患者人数约5 000万,预计到2050年,全球AD患者人数将达到1.52亿[2]。而我国的形势更为严峻,2014年我国约有600万AD患者,总数量已居世界第一。同时也是世界上患病人数增长最快的国家之一。由此可见,我国已成为AD的重灾区。AD已给世界上数以千万计的患者造成了极大的痛苦,同时对社会和患者家庭带来了巨大的压力。因此,开发快速、简便和准确的淀粉样β-蛋白质(Amyloid-β proteins,Aβ)聚集抑制剂筛选方法,对于缓解或治疗该类疾病至关重要。
淀粉样假说认为AD的发生发展是由Aβ的错误折叠和聚集引起的。而Aβ是由β-分泌酶和γ-分泌酶依次水解淀粉样前体蛋白产生的含有39~43个氨基酸的短肽。虽然Aβ的生理功能未知,但大量研究表明健康人群和AD患者的体内均含有Aβ。最大的差别在于它们存在的量和聚集形态不同。游离的Aβ没有毒性作用,而错误折叠和聚集形成的富含β-折叠结构的聚集体才会导致神经元死亡,并最终引起AD。因此,研究Aβ错误折叠和聚集的分子机制对于阐明AD的病理学机制具有非常重要的作用。上述研究的前提是要获得大量具有生物活性和高纯度的Aβ。由于Aβ在人体内含量极少,且其自身极易聚集的特性决定了Aβ难于从人体中提取获得。目前,对于Aβ这种分子量较小的短肽,主要利用现有的短肽固相合成技术获得。除了化学合成技术,基于分子生物学技术的外源表达是获得目标蛋白质的又一常用方法。尤其是随着近年来基因工程和合成生物学技术的快速发展,越来越多的生物大分子利用异源表达技术获得,然后利用蛋白质分离纯化技术获得高纯度的目标蛋白。微生物表达技术除了获得淀粉样蛋白质之外,该技术还常用于基于微生物体系的体内筛选技术的建立。结合本课题组的相关研究结果,本文对微生物技术在Aβ生物表达和抑制剂筛选系统的构建中的应用进行了综述。
1 Aβ的微生物表达
Aβ的错误折叠和聚集与AD的发生发展密切相关。Aβ聚集特性的研究和聚集抑制剂的开发均需要大量高纯度的Aβ。虽然利用现有的固相合成短肽技术完全能够合成Aβ,但由于Aβ固有的强疏水特性,会使化学合成的Aβ含有大量的杂质,降低了其纯度,严重影响其聚集特性及聚集体的细胞毒性。此外,Finder等[3]研究发现微生物表达的Aβ42聚集速度比化学合成的要快,且其聚集体的细胞毒性也更强。因此,利用基因工程技术来生物表达Aβ就成为近年来的研究热点之一[4-5]。
一般情况下,通过基因工程方法在大肠埃希菌中表达的蛋白质N端通常会含有一个甲硫氨酸M。同样,单独在大肠埃希菌中表达Aβ后同样会获得N端带有一个多余残基M的Aβ变异蛋白,为了方便描述将其命名为MAβ。例如,Walsh等[6]利用此方法在大肠埃希菌中表达获得了MAβ40和MAβ42包涵体。将包涵体收集后经尿素溶解和离子交换色谱法纯化后,蛋白产量为20~35 mg/L发酵液。总的来说,本方法所用实验设备及操作手段均较为简单,成本不高,且目标蛋白产量较高,为大量获得MAβ提供了借鉴。由于Aβ单独表达时常以包涵体形式存在,因此后期的包涵体蛋白的纯化和复性方法极其关键。考虑到Aβ的分子量较小,没有固定的二级结构且其构象转换非常迅速,因此Aβ的复性就没有任何意义。因此MAβ的诱导表达和包涵体的纯化就成为制约其应用的主要限速步骤。Hoarau等[7]利用包涵体染色技术,优化了MAβ的诱导表达条件,通过不同截留直径的超滤管对目标蛋白进行粗提纯,再经凝胶过滤层析获得纯度达95%左右的MAβ蛋白。由于超滤过程的损失较大,从而MAβ的产量较低,仅为4 mg/L发酵液,因此还需进一步开发更加高效的表达纯化方法。
综上所述,MAβ的分离是制约其生物表达的主要原因之一。此外,虽然有研究证明N端的多余残基M不会影响体外Aβ的聚集和聚集体的毒性,但与AD的致病蛋白相比多了一个残基。为获得不含有多余残基的Aβ,常见的做法是在Aβ的N端添加酶切位点和一些具有特殊亲和分离功能的标签。其中,组氨酸(His6)标签是微生物表达蛋白质应用最广泛的融合标签之一。例如,Liao等[8]在大肠埃希菌中表达了一种N-端带有His6标签的Aβ42。该His6-Aβ42蛋白经Ni2+柱纯化后,利用TEV蛋白酶将N端的His6标签去除,最后利用高效液相色谱分离纯化Aβ蛋白,最终目标蛋白的纯度可达到95%以上,但其产量仅约为3.5 mg/L发酵液。其产量低是由于要经过酶切和多步纯化,此过程会造成目标蛋白的较大损失。若能省略酶切去除His6标签,则会大大提高Aβ的产量。基于此,我们开发了C端添加His6标签的Aβ变异体——Aβ42-His6[9]。经SDS-聚丙烯酰胺凝胶电泳、点免疫共沉淀反应和MALDI TOF/TOF质谱鉴定,确定产物为Aβ42-His6。利用Nanodrop检测产量为23.0 mg/L,其产量远高于现有的生物表达方法获得的Aβ42。ThT荧光检测结果表明,Aβ42-His6具有比化学合成Aβ42更强的聚集特性。原子力显微镜 (AFM) 检测和圆二色光谱(CD)结果表明,Aβ42-His6具有典型的淀粉样纤维形态并富含β-折叠结构。利用PC12细胞对其诱导的细胞毒性进行检测,发现该变异体多肽具有比化学合成Aβ42更强的细胞毒性。以上结果表明, His6标签的添加不会影响Aβ42的聚集性能和细胞毒性。同时,利用等温滴定量热仪检测了Aβ42-His6与Zn2+之间的相互作用,结果显示Zn2+与Aβ42-His6之间的结合力比与学合成Aβ42间的结合力略强。另外,利用分子动力学模拟(MD)方法发现His6标签对Aβ42自身的二级结构、分子间相互作用力等影响极小。因此,本研究所获得的重组Aβ42-His6可广泛应用于研究其聚集特性和筛选聚集抑制剂等方面。鉴于组氨酸标签及其他标签在蛋白纯化过程中的重要作用,本研究结果对其他淀粉样蛋白质的表达纯化具有重要的指导意义。
综合上述结果发现,利用His6标签获得的目标蛋白部分以包涵体形式存在,给后期蛋白纯化带来诸多不便。为了克服上述包涵体纯化方法带来的问题,一些可溶性融合标签蛋白质也被应用于Aβ蛋白的表达。将Aβ和融合性标签蛋白之间加个Linker,同时在Aβ和上述肽段之间添加一个酶切位点。待分离纯化获得融合蛋白后,再利用特异性酶酶切获得Aβ。麦芽糖结合蛋白(MBP)和谷胱甘肽巯基转移酶(GST)是最常见的两种融合蛋白。 Hortschansky等[10]利用MBP标签,在大肠埃希菌中与Aβ40融合表达。经TEV蛋白酶切除MBP标签后,获得不含多余氨基酸残基的Aβ40,每75 g菌体(湿重)可获得目标蛋白的产量约为50~60 mg。经ThT荧光染色、红外光谱等检测,表明该重组蛋白的聚集受随机成核现象的影响。我们也利用MBP标签实现了Aβ42在大肠埃希菌中的可溶性表达[11]。在该研究中,我们在MBP标签和Aβ42之间加入了一段刚性短肽 (NANP)3和一个高效的TEV蛋白酶切位点(ENLYFQ)。然后优化了蛋白酶切条件,最终获得不含任何多余氨基酸残基的Aβ42,产量约为18 mg/L发酵液,远高于相关研究。
除了MBP标签,GST也是最常见的融合蛋白表达标签之一。Zhang等[12]利用GST标签融合Aβ42在大肠埃希菌中成功可溶性表达了GST-Aβ42融合蛋白。经还原性谷胱甘肽纯化后得到GST-Aβ42。GST标签经凝血酶切除,最终通过高效液相色谱分离获得Aβ42。ThT荧光实验和毒理学实验证明该重组多肽具有良好的聚集特性和细胞毒性。然而本研究最大的问题是产量极低,仅为0.36 mg/L发酵液。其主要原因是GSH纯化柱的纯化效率(约为20%)和凝血酶酶切效率(5%)均很低。另外,Long等[13]发现利用GST标签融合表达Aβ也可能以包涵体形式表达。
利用如MBP和GST等可溶性标签表达纯化Aβ的方法同样也存在以下缺点:去除标签的过程耗时且效率较低,同时极易引入其他杂质;一些酶制剂的价格比较昂贵,增加了成本。因此,不需要去除标签即可获得淀粉样蛋白的方法可能效率更高。ZAβ3是一种结合蛋白,具有一定的Aβ聚集抑制作用。Macao等[14]利用pACYCDuet-1质粒串联表达了ZAβ3和Aβ。表达及纯化过程中ZAβ3的聚集抑制作用,减少了Aβ因聚集而造成的产量损失。最终通过凝胶过滤层析将两者分离,获得较纯的Aβ单体。另外,颜炜群等[15]尝试利用酵母表达系统来异源表达Aβ。但与大肠埃希菌表达系统相比,其表达量和纯度等方面均没有明显地提高。此外,酵母的发酵周期较长,因此该表达系统也并不适合用于Aβ的大量制备。综上所述,目前大肠埃希菌还是异源表达Aβ的常用表达系统之一。
2 淀粉样蛋白质聚集抑制剂的筛选系统
基于上述描述,淀粉样蛋白质的错误折叠和聚集是引起蛋白质构象病的主要原因。因此,开发淀粉样蛋白质聚集的有效抑制剂是治疗上述疾病的有效手段之一。近年来的大量研究发现微生物原核细胞能提供一个简单而强大的生物系统来研究体内淀粉样蛋白聚集的分子机制,为更好地理解与构象疾病相关的蛋白质聚集沉淀开辟了新的视野。同时,这些令人兴奋的证据也为利用细菌细胞筛选具有潜在抗聚集活性的化合物提供了可能。在过去的几年里,已经提出了几种体外和细胞内的方法来开发淀粉样蛋白聚集抑制剂。由于这些新方法具有快速、简便、廉价、重复性好等特点,作为一种寻找抗淀粉样蛋白聚集药物治疗构象性疾病的初步筛选方法引起了人们的极大兴趣。
根据淀粉样蛋白质聚集环境的不同,可以将淀粉样蛋白质聚集抑制剂的筛选方法分为以下两种:体外(invitro)和细胞内(incellular)筛选,体外筛选方法具有快速、便宜和可控性强等优点,但其缺点是与生物体内淀粉样蛋白质聚集的环境差距较大。利用小鼠等哺乳动物进行体内实验来验证抑制剂的效果是必不可少的步骤之一,但使用哺乳动物的体内试验的主要缺点是实验周期太长(从数月到数年),此外还会消耗大量化合物。这些均使哺乳动物体内筛选变得非常昂贵。因此,使用转基因动物进行体内实验通常仅限于选择数量较少的有希望的化合物。可见利用体内筛选方法来筛选大型药物库是不可行的。目前,为了克服利用哺乳动物进行体内筛选所存在的限制,一些更简单的动物模型(非哺乳动物)如斑马鱼、果蝇或线虫等,已广泛应用于淀粉样蛋白质聚集抑制剂的筛选和验证中。此外,细菌(简单的原核生物)和酵母(简单的真核生物)可以成为替代模型,从而实现更加快速简单的从数量巨大的潜在化合物库中筛选出有效的化合物。虽然这些简单的动物模型不能完全取代哺乳动物的主导地位,但它们更便宜、周期更短。因此,可以根据所选择的化合物库的容量来选择不同的筛选方法,最后将筛选获得的性能优异的少数分子在哺乳动物体内或人体内进行验证。
2.1 体外筛选系统
目前,硫黄素T (Thioflavine T, ThT)荧光检测方法是体外鉴定淀粉样蛋白聚集的黄金方法[16]。ThT是一种苯并噻唑染料,在与富含β-折叠结构的淀粉体连接时可提高荧光效率,鉴于此来定性或定量观察淀粉样蛋白的聚集,进一步分析候选化合物的抑制效果。然而,由于许多化合物是发色的,甚至具有内在荧光,因此该方法存在假阳性高等缺陷[17-19]。同时还存在工作量大和效率低等问题。此外,常见的方法还有浊度法、CD、透射电子显微镜(TEM)[20]和AFM[21]等,但操作繁琐,且检测信号往往受样品中共存组分的干扰等[19]。另外,由于体内外生理环境的巨大差异,体外检测获得的抑制剂分子在体内可能难以发挥有效的聚集抑制作用[22-23]。更重要的是,体外方法需要大量高纯度的蛋白[24-25]。这需要在体内表达、纯化或采用昂贵的化学合成方法获得所需蛋白质。这些问题导致在体外筛选潜在的抑制聚集的小分子时,需要耗费大量人力和昂贵的程序来获得足够数量的处于所需聚集状态的纯蛋白质。因此,综合上述方法,可以表明体外筛选实验存在假阳性高、灵敏度低和工作量大等不足。
本课题组针对上述方法存在的问题,基于四苯乙烯基衍生物EPB具有在聚集状态下发光,游离状态下不发光的特征成功构建了EPB-Aβ42和EPB-αSN体外筛选体系。该体系在淀粉样蛋白聚集时发光,不聚集或聚集被抑制时不发光,从而降低了背景荧光的干扰,提高了检测的灵敏度。通过已知的淀粉样蛋白聚集抑制剂EGCG对该体系进行了验证,并利用该体系筛选出了托卡朋、维生素B12和核黄素等具有较好抑制作用的小分子。同时,该体系可以考虑应用于淀粉样蛋白聚集抑制剂的体外高通量筛选。
2.2 细胞内筛选系统
细胞内实验允许研究者在不需要高纯度蛋白的前提下探测化合物对目标淀粉样蛋白的影响[26]。此外,相对于体外实验来说,细胞内系统提供了一个更接近生理的环境来评估淀粉样蛋白的聚集特性及其相关的抑制剂开发[27-28]。由于淀粉样蛋白质在原核和真核生物体内的聚集特性与其生理特性类似[29],因此利用简单的单细胞微生物(如细菌[30-32]和酵母[33-35]等)来筛选淀粉样蛋白质聚集抑制剂是可行的。有报道指出将易于错误折叠和聚集的蛋白质基因插入到报告蛋白的合适位置,报告蛋白的活性将依赖于插入蛋白的错误折叠或聚集状态[36]。近年来,越来越多的报告蛋白由于其自身性质的优越性,在监测与神经退行性疾病相关的蛋白质错误折叠方面得到广泛应用,为抑制剂的开发提供了新的方法。常见的报告蛋白包括绿色荧光蛋白 (GFP)[37]和β-内酰胺酶(βlac)[25]等。Kim等[38]利用GFP在大肠埃希菌体内构建了Aβ42-GFP融合体系(图1),由于上游Aβ42序列的聚集和/或不溶性阻止下游GFP的正确折叠和随后的荧光,所以该体系在大肠埃希菌中表达后不发光。然而,添加能够抑制Aβ42聚集的化合物后,能够恢复GFP的折叠,进而通过荧光的变化进行化合物的筛选。然而值得注意的是,GFP分子量一般较大,与淀粉样蛋白偶联可能会对淀粉样蛋白的聚集特性造成影响。此外,游离的荧光蛋白同样发光,造成较大的背景荧光。因此,以上问题限制了该方法在筛选淀粉样蛋白聚集抑制剂中的应用。
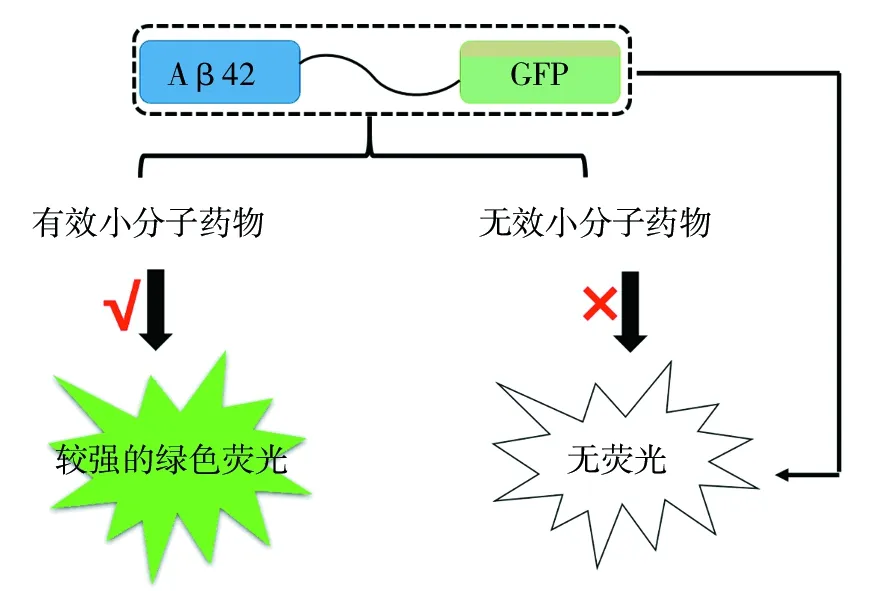
图1 Aβ42-GFP融合体系示意图Fig.1 Schematic diagram of the fusion system Aβ42-GFP
此外,另一种常用的报告蛋白是TEM1-β-内酰胺酶(TEM1-β-lactamase, βlac),其位于革兰阴性菌的细胞周质中。利用它构建的βlac三联体融合体系能将蛋白质稳定性与宿主菌对抗生素的抗性有效联系起来[39]。目前,基于βlac的三联体融合体系目前已广泛应用于优化蛋白质的体内折叠和体内识别小分子聚集抑制剂等[25,40-41]。近期,我们也在βlac的结构基础上将Aβ42插入到βlac 196和197氨基酸残基之间,构建了BL21-βlac-Aβ42重组菌株(图2)。并通过斑点滴定实验、平板涂布实验和菌体浓度变化检测等实验方法,利用已知的Aβ42聚集抑制剂EGCG、固绿和姜黄素对该体系进行了验证。该体系的特点是:基于βlac水解氨苄青霉素的特性,通过观察菌落的生长状况和允许菌落生长的最大细胞稀释倍数等指标来直观评估待测化合物对Aβ42聚集的影响。避免了体外筛选方法存在的不足,同时有望成为体内高通量筛选的新方法。为淀粉样蛋白聚集抑制剂的筛选提供新的方向。
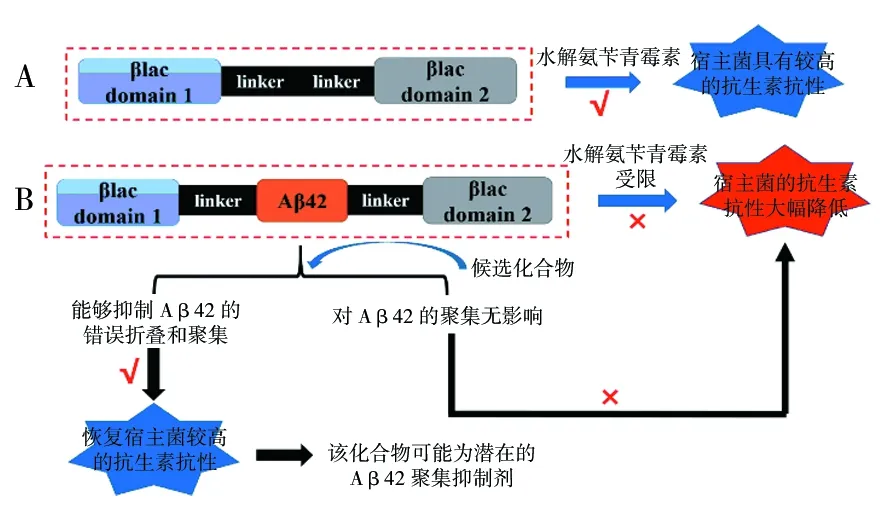
图2 βlac-Aβ42融合体系示意图Fig.2 Schematic diagram of the fusion system βlac-Aβ42
3 展 望
越来越多的研究证明,Aβ的错误折叠和聚集对AD的发生和发展至关重要。因此对于Aβ的结构功能、理化性质和聚集特性等方面的研究以及开发有效的聚集抑制剂成为新的研究热点。但这均需要大量高纯度Aβ。目前淀粉样蛋白的获得主要有固相化学合成、从组织细胞中提取和生物表达纯化等,然而由于淀粉样蛋白自身的高疏水性,对大量获得目标蛋白产生了较大的阻碍。近年来,随着生物工程技术,尤其是分子生物学技术的不断发展,人们对微生物表达系统的研究和认识越来越深入,利用大肠埃希菌表达系统来达到淀粉样蛋白的大量表达和纯化已受到广泛关注。本文详细综述了以大肠埃希菌为表达系统的Aβ的表达和纯化,为合理选择表达工具和方案设计有一定的借鉴意义。
然而,体外获得的Aβ在性质研究和抑制剂筛选方面存在很大缺陷,因为其与体内的聚集过程仍然存在较大的差异。同时利用该表达系统表达外源蛋白易形成包涵体,增加了分离纯化工作量,同时变性复性的过程对蛋白性质也会产生不良影响。因此开发更加高效、便捷的表达体系或表达工具,亟待全世界研究人员的共同努力。此外,目前用于研究蛋白质聚集及其抑制的体外技术是昂贵和耗时的。本文同时介绍了大肠埃希菌体内Aβ聚集抑制剂筛选系统的构建和应用,其为筛选有效的Aβ聚集抑制剂提供了新的见解,为AD治疗指明了新的方向。
猜你喜欢
当代医药论丛(2022年1期)2022-01-27
文萃报·周五版(2021年34期)2021-09-13
天津医科大学学报(2021年4期)2021-08-21
昆明医科大学学报(2021年8期)2021-08-13
浙江临床医学(2021年3期)2021-04-25
现代临床医学(2021年2期)2021-03-29
临床医药文献杂志(电子版)(2020年80期)2021-01-20
爱你·健康读本(2020年8期)2020-08-23
爱你(2020年22期)2020-08-18
安徽医科大学学报(2015年9期)2015-12-16
