
血管紧张素-(1-7)通过调控ClC-3氯离子通道及Nec对抗高糖诱导的内皮细胞损伤
2019-11-11 05:16程飞李诗成陈景福李忠荣赖国华涂昌兰军
广州医科大学学报 2019年3期
程飞,李诗成,陈景福,李忠荣,赖国华,涂昌,兰军
(东莞市第三人民医院心血管内科,东莞市心血管病研究所,广东 东莞 523326)
血管紧张素-(1-7)[Ang-(1-7)]由血管紧张素转换酶 2(ACE2)降解 Ang-Ⅱ而生成,是肾素-血管紧张素系统(RAS)的一个重要成员,具有对抗 Ang-Ⅱ的作用。肾素-血管紧张素系统(renin-antiotensin system,RAS)在心血管疾病及代谢性疾病的发生中起重要的作用。最近有报道指出,糖尿病患者血浆Ang-(1-7)水平降低[1],ACE2表达减少;ACE2过表达的糖尿病小鼠可改善血糖调控[2];Ang-(1-7)的非肽类似物AVE-099可减轻糖尿病引起的心血管损害[3]。在敲除Mas受体基因的小鼠,对胰岛素的敏感性和葡萄糖耐受性降低。而且,在转基因的大鼠,通过过表达Ang-(1-7)则能改善脂质和糖代谢[4]。这些研究表明了Ang-(1-7)可能在糖尿病患者中的心血管并发症中扮演着重要的作用。近年来研究也表明了,Ang-(1-7)能保护血管内皮细胞对抗高糖诱导的损伤作用[5-7],而其机制仍未完全明确。
ClC-3氯通道属于电压依赖性氯通道家族,普遍存在于哺乳动物细胞中可作为容积调节性氯通道或Cl-/H+交换体参与调节细胞的PH、容积、增殖、分化、迁移、凋亡等生理病理活动。ClC-3氯通道于血管内皮细胞功能关系密切。与其参与血管内皮细胞的氧化应激、炎症反应等密切相关[8]。但是,ClC-3氯通道是否参与高糖引起血管内皮细胞损伤及其作用机制尚未明确。
有研究者发现一种非凋亡性程序性死亡方式,具有规律性的调控机制,参与缺血性心脏血管疾病的发生,这种方式被命名为:坏死性凋亡(necroptosis,Nec)。Degterev等将Nec定义为:一种与坏死具有相似的形态学特征(包括包膜完整性破坏,细胞体积及胞内细胞器肿胀),但其细胞死亡方式为可调控的非caspase依赖性的程序性细胞死亡,即在caspase抑制的条件下,死亡受体与配体的结合可触发坏死性凋亡。对Nec的研究是目前国际上极其活跃的前沿热点。不断增加的证据表明:Nec的诱导与阻截涉及机体自身稳定、正常发育及多种疾病的发展与变化。Nec的特异性阻断剂ncerostatin-1(Nec-1)可通过谷胱甘肽(GSH)、凋亡诱导因子和腺病毒E1B 19 kD-相关作用蛋白3(BNIP3)相关通路抑制谷氨酸介导的细胞损伤。近来研究表明,Nec与高糖诱导血管内皮细胞损伤密切相关[9]。然而,Nec参与高糖引起血管内皮细胞损伤及其作用机制需要进一步探讨。
为此,本研究建立高糖损伤人脐静脉血管内皮细胞模型,旨在探讨: ( 1) ClC-3在高糖诱导血管内皮细胞损伤中的作用机制; (2) Nec在高糖诱导血管内皮细胞损伤中的作用机制;(3)Ang-(1-7) 预处理能否通过通过调控ClC-3氯离子通道及Nec对抗高糖诱导的内皮细胞损伤。
1 材料与方法
1.1 材料与试剂
血管紧张素-(1-7)、NPPB、Nec-1、Hoechst33258和DCFH-DA购自Sigma; CCK-8试剂盒购自Dojindo; DMEM(低糖)培养基购自HyClone;胎牛血清(fetalbovine serum,FBS)购自 Gibco; ClC-3抗体、RIP3抗体和GAPDH 抗体购自 Proteintech; 人脐静脉内皮细胞(HUVECs) 购自广州吉妮欧公司。
1.2 细胞培养
HUVEC的体外培养细胞培养在含10% FBS的低糖DMEM培养基中,将HUVECs置于5% CO2、37 ℃的温箱中培养。待HUVECs融合达80%后,用0.25% 胰蛋白酶-EDTA消化HUVECs,适度消化后加入完全培养基终止胰蛋白酶的消化作用。用灭菌枪头将瓶壁细胞吹落形成细胞悬液。于1200 r/min离心5min,弃上清,按1∶ 3传代HUVECs。实验前换用无血清的DMEM培养液培养HUVECs 12 h,然后进行分组实验。
1.3 实验分组
实验分为8组: (1)正常对照(control)组; (2)高糖(highglucose,HG) 组: 40 mmol/L葡萄糖处理HUVECs 24 h; (3) Ang-(1-7)+HG组: 2 μmol/L Ang-(1-7)处理HUVECs 30 min,撤去培养液后用PBS洗3次,接着应用40 mmol/L葡萄糖处理HUVECs 24 h; (4) NPPB+ HG 组: 20 μmol/L NPPB处理HUVECs 30 min,撤去培养液后用PBS洗3次,再用40 mmol/L葡萄糖作用HUVECs 24 h; (5)Nec-1+HG 组: 10 μmol/L Nec-1预处理HUVECs 30 min,撤去培养液后用PBS洗3次,再用 40 mmol/L葡萄糖作用24 h; (6) Ang-(1-7)组: 2 μmol/L Ang-(1-7)作用于HUVECs 30 min,撤去培养液后用PBS洗3次,再用DMEM培养基作用HUVECs 24 h; (7) NPPB组: 20 μmol/L NPPB作用于HUVECs 30 min,撤去培养液后用PBS洗3次,再用DMEM培养基作用24 h; (8)Nec-1组:10 μmol/LNec-1作用于HUVECs 30 min,撤去培养液后用PBS洗3次,再用DMEM培养基处理HUVECs 24 h 。
1.4 CCK-8法实验测定HUVECs存活率
将HUVECs接种于96孔培养板中,待HUVECs 在培养孔内生长至约80% 时,按照实验要求给予不同处理,弃培养液,用预冷的PBS洗3次,于每孔中加入10 μL CCK-8溶液,37 ℃孵育24 h,使用酶标仪检测各孔吸光度(A),波长设定为450 nm。按公式:HUVECs存活率(%) =处理组A/对照组A×100%,求出处理组细胞存活率,实验重复5次。
1.5 Western blot法检测ClC-3和RIP3的表达
将HUVECs接种于60 mm培养皿中,待HUVECs贴壁生长约80%时给予不同处理因素,弃培养液,用预冷的PBS洗3次,各皿加入100 μL 裂解液后置4 ℃裂解40 min,于12 000 r/min离心15 min,取上清,蛋白浓度采用BCA法进行测定。总蛋白经SDS-PAGE分离后,转移到PVDF膜上。用5%脱脂奶粉封闭90 min,分别加入各种I抗[ClC-3和RIP3(1∶2500)],4℃过夜,然后用TBST洗5次(每次10 min),加入II抗(1∶10000),室温孵育60 min,然后再用TBST洗5次(每次10 min)。ECL发光液将PVDF膜显色,暗室曝光,凝胶成像系统扫描分析结果。
1.6 SOD活性的检测
将HUVECs接种于60 mm培养皿中,待HUVECs贴壁生长约80%时给予不同处理因素,弃培养液,用预冷的PBS洗3次,各皿加入100 μL裂解液后置4 ℃裂解40 min,于12 000 r/min离15 min,取上清,采用BCA法进行蛋白定量,再将各样品的总蛋白量调至相同。然后根据“Dojindo Lab. Japan”提供的SOD活性试剂盒说明书的操作步骤,检测SOD对超氧化物的抑制率。
1.7 细胞内活性氧簇 (reactive oxygen species,ROS)含量的检测
DCFH-DA是一种自身不能发荧光的化合物,可自由穿过细胞膜,被胞内 ROS氧化为有荧光的DCF。通过检测DCF的荧光强度即可反映细胞内ROS水平。将HUVECs接种于6孔培养板中,当细胞生长贴壁生长至80%时,给予各实验组不同的处理因素后,弃培养液,PBS冲洗3次,将HUVECs与10 μmol/L DCFH-DA于37 ℃孵育30 min,再用PBS冲洗3次。在荧光显微镜下随机地选取5个不重复区摄片,采用ImageJ 1.41软件分析各视野平均绿色荧光强度,进而对每组的各个样本进行统计分析。
1.8 线粒体膜电位的检测
将HUVECs接种于96孔培养板中,待HUVECs在培养孔内生长至约80%时,按照实验要求给予不同处理,弃培养液,用预冷的PBS洗3次,用10 μg/L Rh123的无血清培养基于37 ℃温箱中孵育60 min,然后用预冷的PBS冲洗3次。在荧光显微镜下随机选取5个不重复区摄片,细胞核周围绿色的亮点即为摄取了Rh123的线粒体。用Image J 1.47i软件分析5个视野绿色荧光强度的平均值,再对每组的各样本进行统计分析。重复5次。
1.9 统计学处理
2 结果
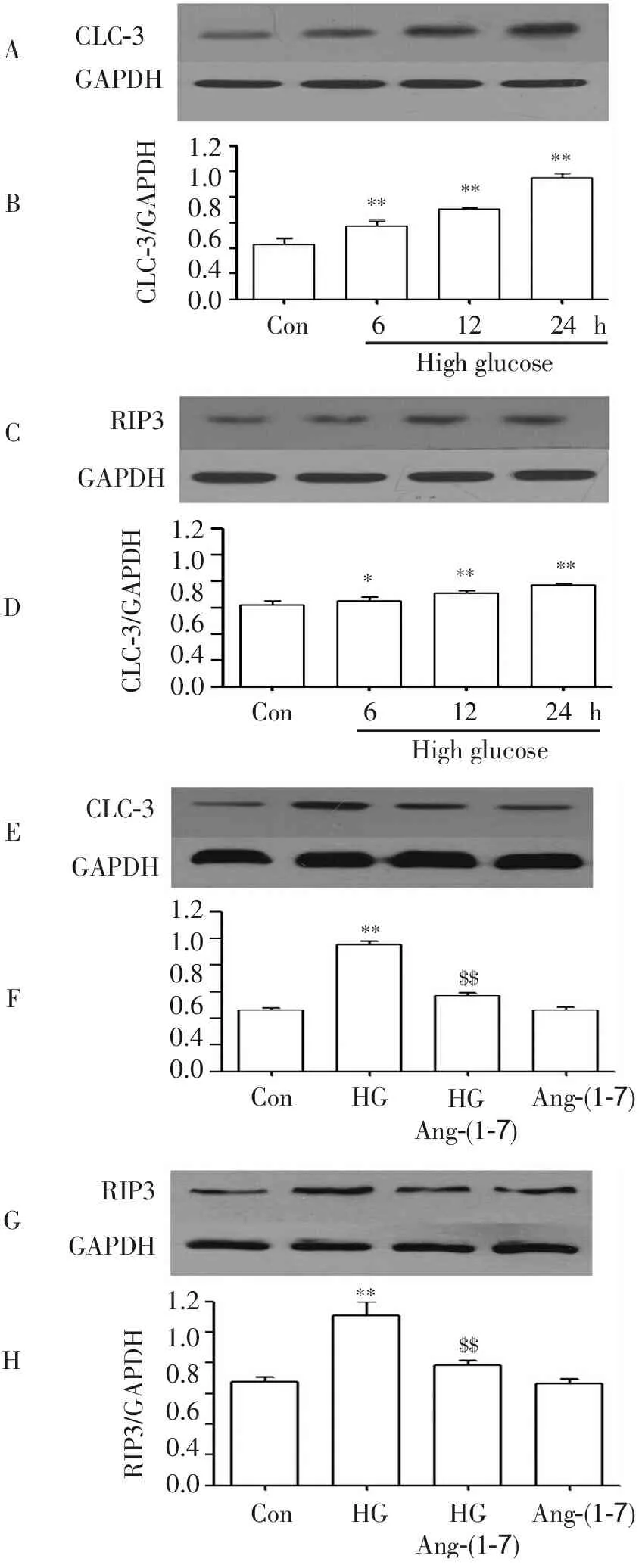
2.1 血管紧张素-(1-7)抑制高糖上调血管内皮细胞ClC-3及RIP3表达水平
采用40 mmol/L葡萄糖分别处理HUVECs不同时间(0,6,12和24 h)后发现,40 mmol/L葡萄糖可显著上调ClC-3及RIP3表达水平。与对照组相比,差异有统计学显著性(P<0.01),其中在24 h时ClC-3及RIP3表达水均达到最高,见图1 A-D。
应用高糖(40mmol/L葡萄糖)处理HUVECs 24 h使ClC-3及RIP3表达水平升高,与对照组相比,差异有统计学显著性(P<0.01)。但是,在高糖处理血管内皮细胞前,应用2 μmol/L Ang-(1-7)预处理30 min使高糖对ClC-3及RIP3表达水平上调作用显著减弱,与HG处理组比较,差异有统计学显著性(P<0.01)。2 μmol/L Ang-(1-7)本身对内皮细胞ClC-3及RIP3表达水平无明显的影响,见图1 E-H。
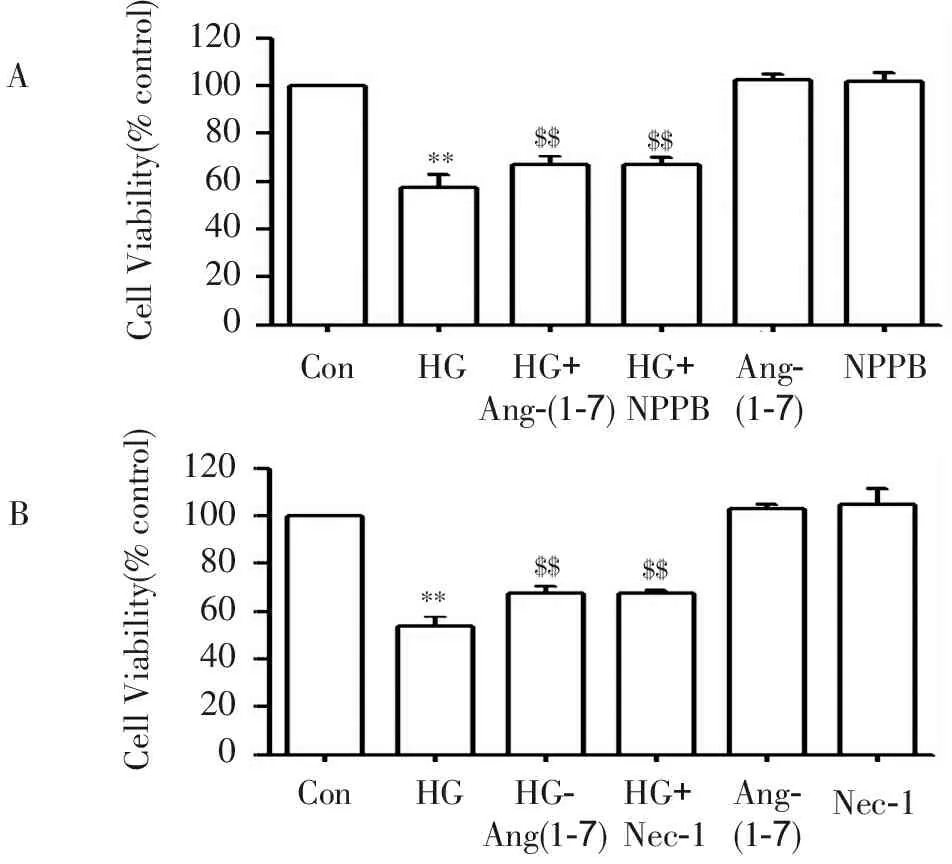
2.2 Ang-(1-7) 、NPPB及Nec-1减轻HG引起血管内皮细胞的心肌细胞毒性
图2显示,采用40 mmol/L葡萄糖处理HUVECs 24 h可以诱发心肌毒性作用,表现为内皮细胞存活率降低,与对照组比较,差异显著(P<0.01)。然而,2μmol·L-1Ang-(1-7)或20 μmol·L-1NPPB(ClC-3抑制剂)或10 μmol·L-1Nec-1与HG共处理心肌细胞24 h可明显地减弱HG引起的心肌细胞毒性,使细胞存活率升高,与HG组分别比较,差异均具有统计学意义(P<0.01)。Ang-(1-7)或 NPPB或Nec-1本身不影响心肌细胞存活率(P>0.05,图2)。
CLC-3GAPDHCLC-3/GAPDH******61224ConHighglucoseh**1.21.00.80.60.40.0CLC-3/GAPDH1.21.00.80.60.40.0RIP3GAPDH***61224ConHighglucoseh**$$CLC-3GAPDH1.21.00.80.60.40.0ConHGHGAng-(1-7)Ang-(1-7)$$ConHG**1.21.00.80.60.40.0GAPDHRIP3RIP3/GAPDHBCDEFGHAAng-(1-7)HGAng-(1-7)
图1 血管紧张素-(1-7)抑制高糖上调血管内皮细胞ClC-3及RIP3表达水平
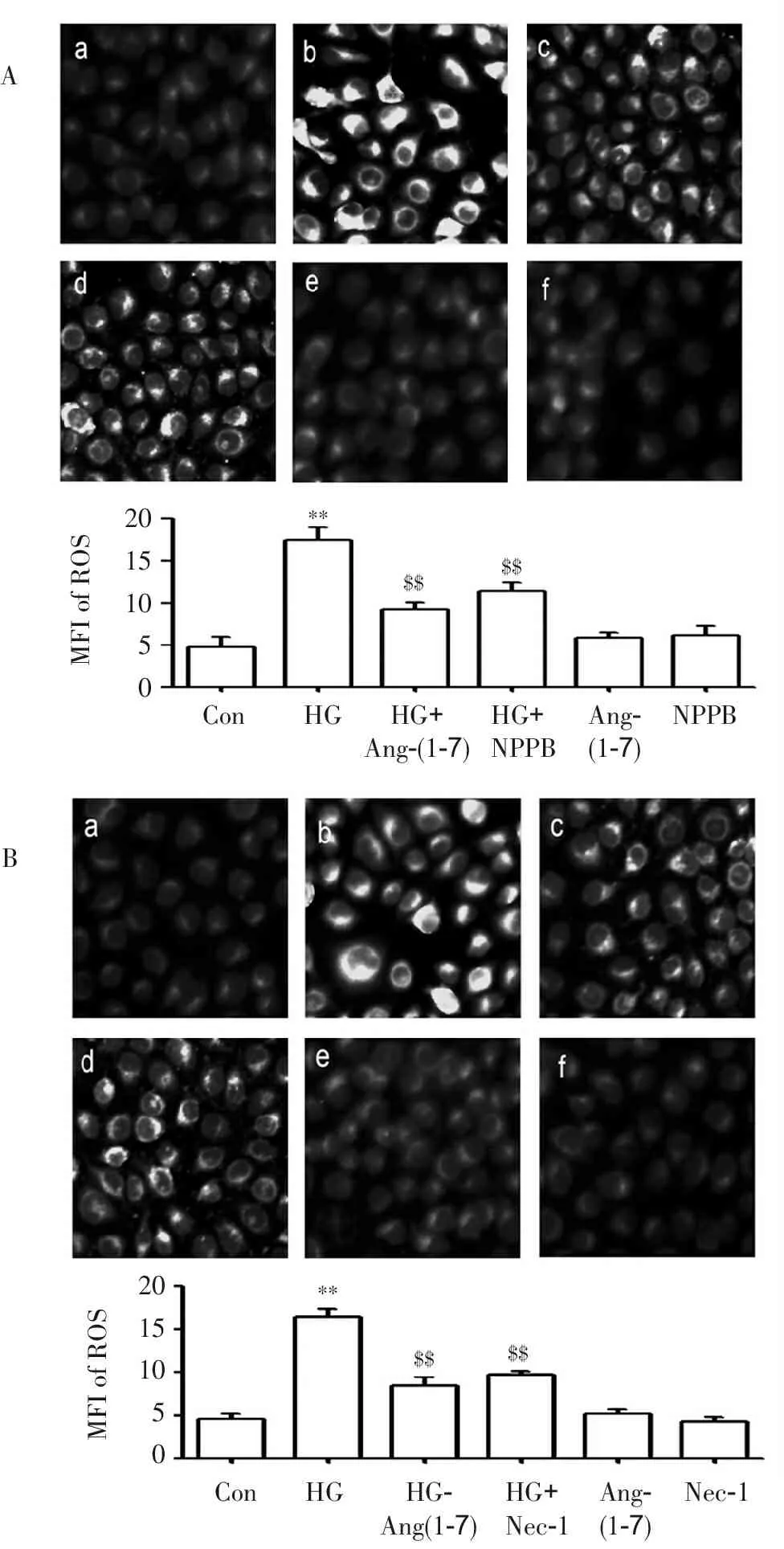
2.3 Ang-(1-7) 、NPPB及Nec-1减轻高糖引起的人脐静脉内皮细胞的氧化应激
HG作用于HUVECs 24 h可使胞内DCFH的平均荧光强度(mean fluorescence intensity,MFI;反映ROS水平的一个指标)明显增强,与正常对照组相比,两组差异有统计学显著性(P<0.01)。但经2 μmol·L-1Ang-(1-7)或20 μmol·L-1NPPB(ClC-3抑制剂)或10 μmol·L-1Nec-1预处理HUVECs 30 min可使内皮细胞内 ROS堆积明显减少,与高糖组相比,差异有统计学显著性(P<0.01)。而单独2 μmol·L-1Ang-(1-7)或20 μmol·L-1NPPB(ClC-3抑制剂)或10 μmol·L-1Nec-1预处理则对胞内ROS生成无明显作用,见图3。
120100806040200CellViability(%control)120100806040200CellViability(%control)ConHGHG-Ang(1-7)Ang-(1-7)HG+Nec-1ConHGHG+Ang-(1-7)Ang-(1-7)HG+NPPBNPPBNec-1**$$$$**$$$$AB
**P<0.01与对照组比较,$$P<0.01与HG组比较。
图2 Ang-(1-7) 、NPPB及Nec-1减轻HG引起血管内皮细胞的心肌细胞毒性
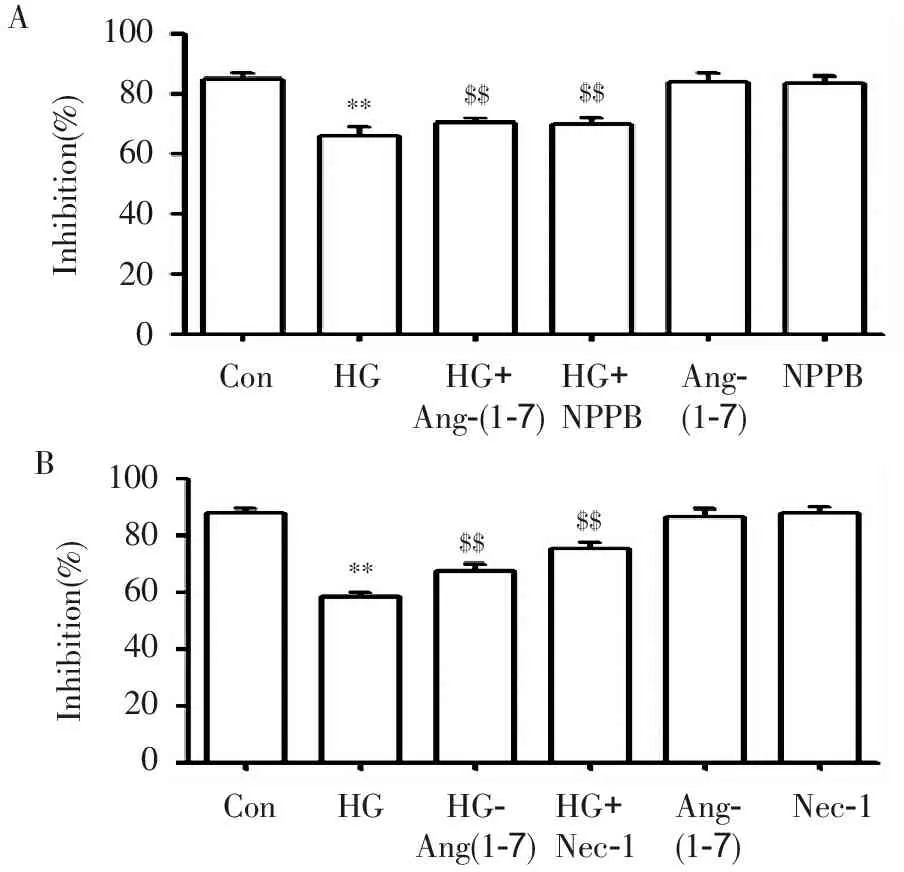
2.4 Ang-(1-7) 、NPPB及Nec-1阻断葡萄糖对血管内皮细胞 SOD的抑制作用
图4结果显示,40mmol/L葡萄糖处理HUVECs 24 h,明显抑制了SOD的活性,Ang-(1-7) 、NPPB及Nec-1本身对SOD活性无明显的影响,但是能阻断40 mmol/L葡萄糖对HUVECs的SOD的抑制作用,使SOD活性明显增强。
2.5 Ang-(1-7) 、NPPB及Nec-1减轻HG诱发的血管内皮细胞MMP丢失
图5显示,HG处理HUVECs 24 h后可使MMP明显降低(图5 Ab、Bb),与对照组(图5 Aa、Ba)比较,差异具有统计学意义(P<0.01)。但是,2 μmol·L-1Ang-(1-7)或 20 μmol·L-1NPPB(ClC-3抑制剂)或10 μmol·L-1Nec-1与HG共处理HUVECs 24 h后可使MMP降低减少,与HG组分别比较,差异显著(P<0.01)。
**$$$$20151050MFIofROSConHGHG+Ang-(1-7)Ang-(1-7)HG+NPPBNPPB**$$$$20151050MFIofROSConHGHG-Ang(1-7)Ang-(1-7)HG+Nec-1Nec-1AB
**P<0.01与对照组比较,$$P<0.01与HG组比较。
图3 Ang-(1-7) 、NPPB及Nec-1减轻HG诱发的血管内皮细胞氧化应激
3 讨论
与我们以往报道相一致[13],本研究在HG处理血管内皮细胞的实验模型,再次证实HG能引起血管内皮细胞出现多种损伤,表现为细胞存活率降低(细胞毒性作用),SOD生成下降和ROS(氧化应激)生成增多及MMP丢失(线粒体受损)。同时,本实验观察到HG能明显促进ClC-3的表达,提示HG能激活血管内皮细胞ClC-3氯离子通道。而ClC-3氯通道于血管内皮细胞功能关系密切。与其参与血管内皮细胞的氧化应激、炎症反应等密切相关[8,11]。
ConHGHG-Ang(1-7)Ang-(1-7)HG+Nec-1Nec-1ConHGHG+Ang-(1-7)Ang-(1-7)HG+NPPBNPPB$$$$**$$$$**100806040200Inhibition(%)100806040200Inhibition(%)AB
**P<0.01与对照组比较,$$P<0.01与HG组比较。
图4 Ang-(1-7) 、NPPB及Nec-1阻断葡萄糖对 SOD的抑制作用
支持本文的研究结果。为了进一步证实ClC-3氯离子通道在HG损伤血管内皮细胞中的作用,我们观察了ClC-3离子通道抑制剂NPPB对血管内皮细胞损伤的影响。研究结果表明,NPPB能显著地减轻HG对血管内皮细胞的多种损伤作用,表现为使细胞存活率升高,SOD生成增加、ROS生成及MMP丢失减少等,清晰地提示ClC-3离子通道介导HG引起的细胞毒性、氧化应激及线粒体损伤等作用。
此外,我们研究小组还观察到,HG能明显促进RIP3的表达,提示HG能激活血管内皮细胞Nec。Liang GZ,等研究表明Nec-1可通过抑制β-catenin通路保护血管内皮细胞作用[8],提示Nec在血管内皮细胞损伤中扮演重要作用。为了验证Nec在HG损伤血管内皮细胞中的作用,我们观察了Nec抑制剂Nec-1对血管内皮细胞损伤的影响。与NPPB作用类似,Nec-1同样能减轻HG对血管内皮细胞的多种损伤作用。我们的研究结果提示Nec介导HG引起的细胞毒性、氧化应激及线粒体损伤等作用。
Ang-(1-7)是肾素-血管紧张素系统中具有与Ang-II相反的多种生理作用,其中研究表明Ang-(1-7)具有血管内皮保护[14]。而在糖尿病导致的心血管并发症中,Ang-(1-7)的血管内皮保护作用使得其成为防治糖尿病心血管损伤的热点之一。既往研究表明,Ang-(1-7)能保护血管内皮细胞对抗高糖诱导的损伤作用[5-7],然而其机制仍未完全明确,Ang-(1-7)能否通过调控ClC-3氯离子通道及Nec对抗高糖诱导的内皮细胞损伤尚未见报道。本研究证实,外源性Ang-(1-7)能抑制HG对ClC-3及RIP3的激活;ClC-3抑制剂NPPB及Nec-1抑制剂Nec-1均能产生类似于Ang-(1-7)的血管内皮细胞保护作用,进一步提示通过抑制ClC-3氯离子通道及Nec可能是Ang-(1-7)保护心肌细胞对抗HG引起的损伤的重要机制之一。由于离体细胞实验的研究结果可能与在体实验的结果存在一些差异,因此,本研究拟在今后在糖尿病动物模型,开展进一步的研究。
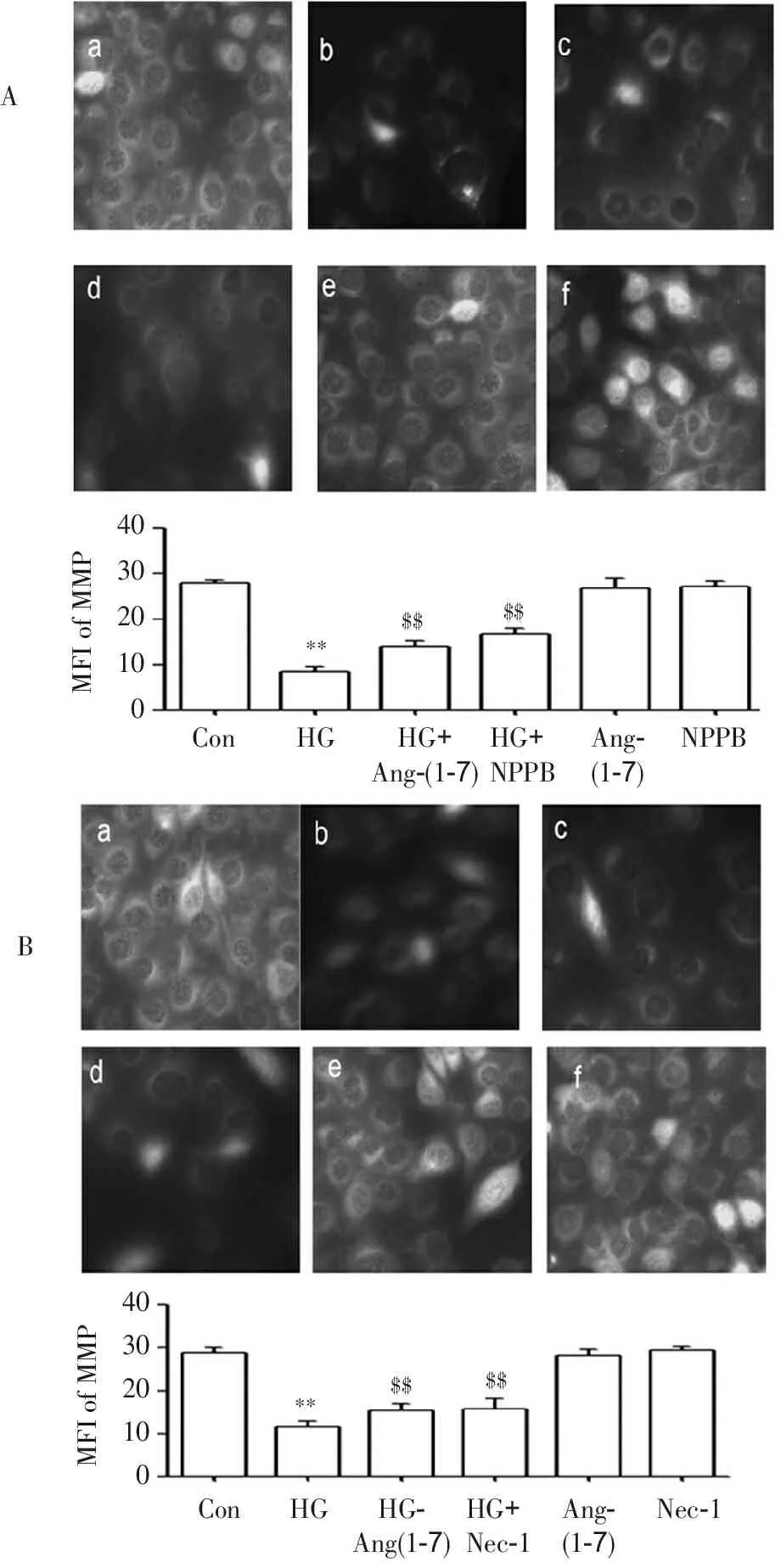
$$**$$$$$$**403020100MFIofMMPConHGHG+Ang-(1-7)Ang-(1-7)HG+NPPBNPPB403020100MFIofMMPConHGHG-Ang(1-7)Ang-(1-7)HG+Nec-1Nec-1AB
**P<0.01与对照组比较,$$P<0.01与HG组比较。
图5 Ang-(1-7) 、NPPB及Nec-1抑制HG引起的HUVECs MMP降低
猜你喜欢
中学生物学(2021年8期)2021-11-02
建材发展导向(2021年11期)2021-07-28
当代水产(2020年10期)2020-03-17
当代水产(2019年8期)2019-10-12
中国果业信息(2019年1期)2019-01-05
中国畜牧兽医文摘(2018年6期)2018-07-28
中成药(2018年6期)2018-07-11
中成药(2017年8期)2017-11-22
科学启蒙(2015年8期)2015-08-07
长江蔬菜(2015年3期)2015-03-11
