
塞来昔布的合成及表征
2019-11-07 11:04:16王福生苏艳华
生物化工 2019年5期
王福生,苏艳华
(瑞阳制药有限公司,山东淄博 256100)
塞来昔布是新一代非甾体镇痛抗炎药,通过选择性抑制环氧化酶-2(COX-2)来抑制前列腺素的合成,从而达到抗炎症、镇痛的效果。当前国内外报道塞来昔布的合成文献[1-3]较多,较为成熟的路线如图1所示。
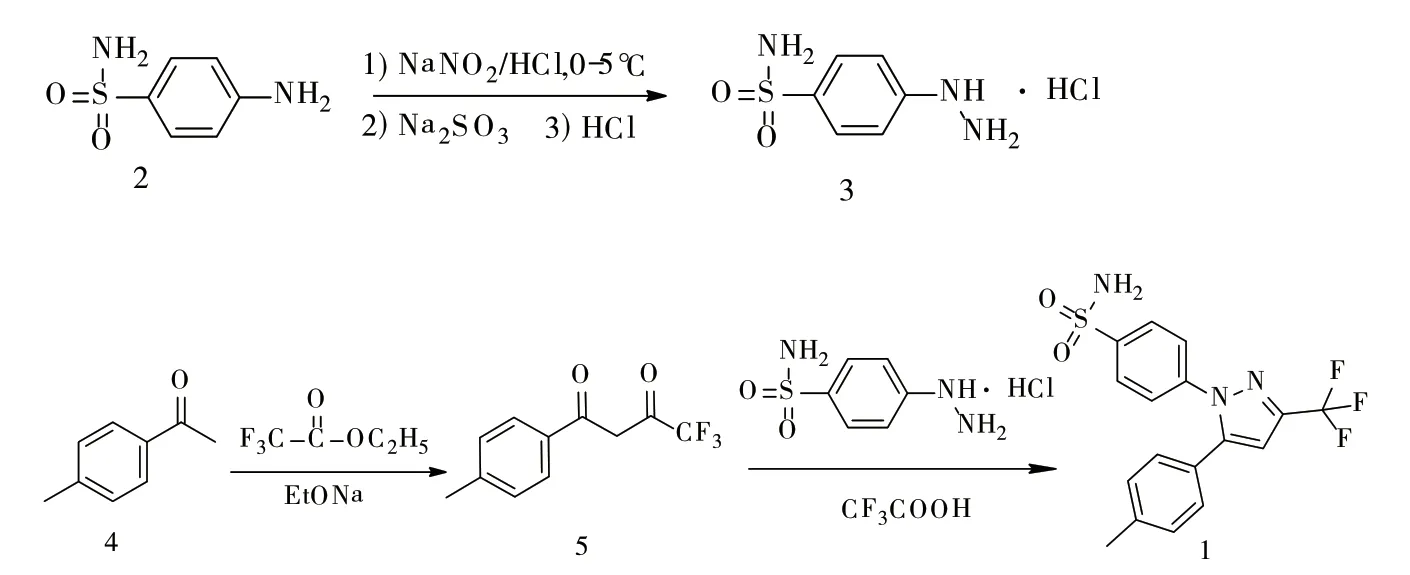
图1 塞来昔布的合成路线
本文在参考相关文献[4-5]后采用上述路线,并对上述路线的具体工艺进行了改进。本文中化合物4采用乙醇钠(乙醇钠溶液含量16%)为缩合剂合成化合物5,合成后的化合物5不经母液分离直接与化合物3反应两步一锅生成化合物1,收率达89.5%,较其他文献[6-8]工艺收率和质量有明显提高。
1 实验部分
1.1 仪器及试剂
Vario EL Ⅲ元素分析仪(Elementar公司);Nexus型傅里叶变换红外(FT-IR)分析仪(Thermo Nicolet公司);Waters SQD2液相色谱质谱联用仪(Waters公司);Avance Ⅲ600MHz核磁共振谱仪(BRUKER公司)。
对氨基苯磺酰胺(AR)、盐酸(AR)、亚硝酸钠(AR)、无水亚硫酸钠(AR)、对甲基苯乙酮(工业级)、三氟乙酸乙酯(AR)、乙醇钠溶液(工业级)、三氟乙酸(AR)、异丙醇(AR)、乙醇(AR)。
1.2 塞来昔布的合成
1.2.1 对肼基苯磺酰胺盐酸盐(化合物3)的合成
2L三口烧瓶中加入盐酸(145g,1.45mol)、对氨基苯磺酰胺(100g,0.58mol)和去离子水300g,搅拌降温至0℃。将配制好的亚硝酸钠溶液(44.07g亚硝酸钠溶于126g水中)缓慢滴加到三口烧瓶中,控温0~5℃,约30 min滴毕,滴完后保温搅拌10 min。
5L三口烧瓶中加入水330g,在搅拌下加入无水亚硫酸钠(182.98g,1.45mol),降温至0℃,再加入碎冰130g,将上述反应液加入此体系中,用水20g冲洗2L三口烧瓶,操作完毕后保温搅拌10 min。然后水浴升温至80℃,加入盐酸(392.97g,3.93mol),溶解澄清后停止搅拌和加热,自然降温至室温后析晶12 h,然后抽滤干燥得到中间体对肼基苯磺酰胺盐酸盐约102.41g,纯度98.92%,收率为78.84%。
1.2.2 塞来昔布(化合物1)的合成
2L三口烧瓶中加入乙醇钠溶液(230.84g,0.56mol),在搅拌下加入三氟乙酸乙酯(80.458g,0.57mol),搅拌5 min后加入对甲基苯乙酮(57.13g,0.43mol)。
水浴加热升温至50℃,保温搅拌2 h,反应液直接用于下一步反应。
5L三口烧瓶中加入对肼基苯磺酰胺盐酸盐(100g,0.45mol)、乙醇221g和水157.59g,搅拌下加入三氟乙酸(50.98g,0.45mol),水浴加热升温。当温度升至50℃时,将上步反应液滴加到此体系中,然后保温搅拌30 min,再加入水203.39g,升温至65℃,再滴加水191.67g,保温搅拌30 min,停止加热,搅拌析晶约3 h后抽滤,使用50%乙醇充分洗涤2次后干燥得到固体约145.3g,纯度99.12%,收率为89.5%。
重结晶:500mL三口烧瓶中,装有搅拌、温度计和冷凝管,将50.0g塞来昔布粗品加入三口烧瓶中,再加入160g异丙醇,加热溶解澄清后,再加入2g活性炭,回流搅拌20min后滤除活性炭,将滤液转至干净反应瓶中,加热回流澄清后,缓慢加入8.0g水,缓慢降温析晶待降至20~25℃后,再析晶6h后抽滤,使用少量冷异丙醇的冲洗后干燥得45.2g,纯度为99.83%,收率为90.4%。
2 结果及讨论
2.1 塞来昔布的元素分析
在制备塞来昔布后,为了进一步确定其结构,对产物进行了C、H、N的元素分析,其结果如表1所示。
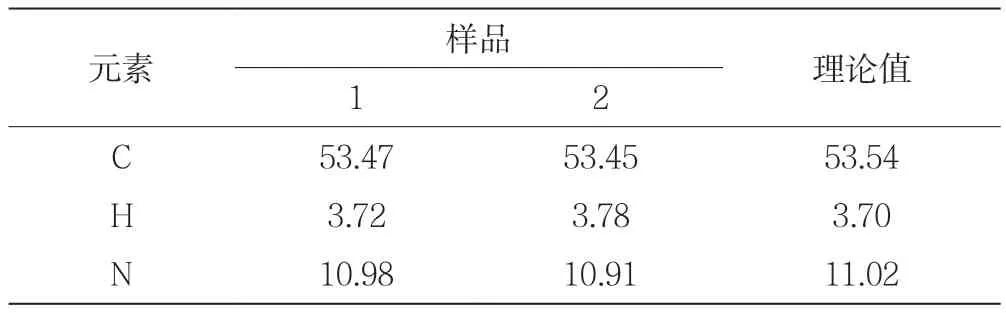
表1 塞来昔布的元素分析
由表1可知,根据样品中C、H和N元素分析结果,各元素实测值平行性良好,实测值与理论值基本一致,为进一步确定其结构进行了IR分析。
2.2 塞来昔布的IR分析
将制备的塞来昔布进行红外光谱分析,其主要吸收峰及归属如表2所示。

表2 塞来昔布的IR谱各吸收峰及归属
由表2可以看出:(1)3098cm-1为苯环中=C-H的伸缩振动吸收,1563cm-1、1499cm-1为苯环骨架的伸缩振动吸收,846cm-1为苯环中=C-H的弯曲振动吸收,说明结构中含有苯环结构;(2)1375cm-1为甲基的C-H弯曲振动吸收,说明结构中含有甲基结构;(3)3339cm-1、3233cm-1为磺酰胺的N-H伸缩振动吸收,1348cm-1为磺酰胺基的SO2的伸缩振动吸收,说明结构中含有磺酰胺结构。
解析结果表明:该样品的红外光谱图特征与目标化合物的化学结构基本相符合。
2.3 塞来昔布LC/MS分析
将制备的塞来昔布进行质谱分析,其分子离子峰及归属如表3所示。

表3 塞来昔布的分子离子峰及归属
由表3可知,质谱测得本品的分子离子峰[M+H]+,其质荷比m/z为382.1,与塞来昔布的分子离子峰(分子量为381.37)一致。
2.4 塞来昔布的1H-NMR分析
将制备的塞来昔布样品进行1H-NMR谱检测,1H-NMR((CD3)2SO,600MHz):δ7.906(d,2H,J=8.4Hz,Ar-H),7.561(s,2H,NH2),7.550(d,2H,Ar-H),7.227(d,2H,Ar-H),7.222(d,2H,Ar-H),7.190(s,1H,吡唑环CH),2.324(s,3H,CH3),综合以上信息,与塞来昔布结构信息吻合。
3 结论
(1)通过元素分析、红外光谱法、质谱法和1H-NMR的检测和表征,确认了塞来昔布目标产物的合成。
(2)本文中化合物4采用乙醇钠(乙醇钠溶液含量16%)为缩合剂合成化合物5,合成后的化合物5不经处理分离直接与化合物3反应两步一锅生成化合物1,收率达89.5%,经精制后纯度达99.83%,该路线总收率为63.8%,经改进后其收率和质量有明显提高。
猜你喜欢
食品工业科技(2021年17期)2021-09-14 00:49:58
高师理科学刊(2020年11期)2021-01-04 08:05:22
理化检验-化学分册(2020年3期)2020-04-24 10:43:44
山东化工(2018年16期)2018-09-12 09:43:38
浙江化工(2017年4期)2017-05-11 02:37:40
中华胃食管反流病电子杂志(2016年3期)2016-10-18 00:45:15
河北医科大学学报(2016年7期)2016-08-12 07:25:19
医学研究杂志(2015年11期)2015-06-10 06:44:03
含能材料(2015年1期)2015-05-10 00:50:52
云南中医学院学报(2014年4期)2014-07-31 18:22:23
