
剪接体蛋白SmD3在果蝇S2细胞中对U2A的影响
2019-10-26 03:24栾晓瑾颜一丹郑倩雯陈万银
医学研究杂志 2019年12期
栾晓瑾 颜一丹 陈 霞 谢 冰 郑倩雯 王 敏 陈万银 乔 晨 于 骏 方 杰
真核生物细胞核内DNA经转录产生未经修饰的前体mRNA(pre-mRNA)后,需要在剪接体(spliceosome)的作用下去除内含子,连接外显子,才能成为成熟的mRNA分子。剪接体由5种细胞核糖核蛋白小体(small nuclear ribonucleoprotein, snRNP)包括U1、U2、U4、U5和U6 snRNPs,以及100多种蛋白质因子组成[1,2]。Sm蛋白是高度保守的RNA结合蛋白家族,真核生物中主要分为经典Sm蛋白和类Sm蛋白(like Sm, LSm),由7种经典Sm蛋白质B/B′、D1、D2、D3、E、F和G所形成的七聚体环是剪接体上与大多数snRNP结合的基本结构,只有U6 snRNP与LSm2-8七聚环结合[3,4]。SmD3作为经典Sm蛋白之一,对于pre-mRNA的剪接至关重要,然而仍不清楚SmD3是否具有剪接因子除外的其他功能[5]。
UAS-Gal4系统是一种转基因技术体系,最早在果蝇中建立,已成功利用Gal4于多种果蝇组织中激活UAS下游靶基因[6]。有研究通过UAS-Gal4系统进行大规模果蝇睾丸RNA干扰(RNAi)筛选,共鉴定了包括SmD3在内的221个参与生殖干细胞自我更新过程的调控因子,具体调控机制尚未明确[7]。近期研究表明,U2 snRNP的关键组成蛋白U2A具有新功能,它是雄性果蝇生育力所必需的,果蝇睾丸生殖细胞缺乏U2A导致未分化的精原细胞积累[8]。尚未有文献报道U2A与SmD3是否存在相互作用。
果蝇S2细胞为果蝇胚胎细胞,由于其基因组可整合多拷贝的质粒,表达外源蛋白的效率较高,已经作为体外实验细胞模型逐渐广泛运用于生物学及医学领域的研究[9,10]。因此,本研究通过SmD3 siRNA及构建pUAS-attB-SmD3表达载体,借助果蝇S2细胞为表达系统实现SmD3表达水平的改变,利用qRT-PCR检测U2A mRNA表达,同时应用免疫荧光法对U2A进行蛋白水平的检测,以探索SmD3对U2A的影响,为研究SmD3的作用机制提供方向。
材料与方法
1.材料:S2细胞从果蝇基因组资源中心获得;Schneider培养基(美国Gibco公司);胎牛血清(以色列Bioind公司);SmD3 CDS 由浙江大学佟超实验组惠赠;U2A抗体由南京医科大学生殖医学国家重点实验室惠赠;带荧光标记的SmD3 siRNA质粒及阴性对照质粒(苏州吉玛基因公司);Xhol Ⅰ、Xbal Ⅰ、TRIzol、反转录试剂盒及qRT-PCR用SYBRGreen染料(日本TaKaRa公司);Oligo-fectamineTM2000试剂(美国Invitrogen公司);Effectene Transfection Reagent(德国Qiagen公司);Opti-MEM(美国Gibco公司);Cy3标记抗兔IgG(H+L)(美国Jackson公司);qRT-PCR引物(上海生工生物公司);Real-timePCR仪:Mx3000P/Mx3005P(美国Agilent公司),荧光显微镜(日本Olympus公司),CO2培养箱(美国赛默飞公司)。
2.构建SmD3过表达克隆:将SmD3 CDS通过聚合酶链反应(PCR)扩增添加Xhol Ⅰ和Xbal Ⅰ限制性内切酶位点。上游引物序列:5′-CCGCTCGAGGTATGTCTATCGGAGTGCCCAT-3′,下游引物序列:5′-GCTCTAGATTTCTACAGGCCGCCGCGTCCT-3′。其反应条件为:95℃、30min,95℃、10s,55℃、30s,72℃、40min,共38个循环;72℃、7min。将PCR产物进行琼脂糖凝胶电泳,割胶回收电泳产物,使用DNA凝胶回收试剂盒对PCR产物予以纯化。用Xhol Ⅰ和Xbal Ⅰ对上步所提取质粒及pUAS-attB表达载体进行双酶切。将酶切产物进行纯化后,将回收的插入片段与载体连接,热转化至DH5α大肠杆菌中,涂布于含有氨苄青霉素的固体培养基上,37℃培养过夜。挑选阳性菌落,提取质粒,所提取质粒用Xhol Ⅰ/Xbal Ⅰ双酶切予以验证,并送予上海生工生物工程公司测序比对。
3.S2细胞培养:S2细胞常规培养采用Schneider培养基,补以10%胎牛血清。S2细胞培养在28℃无CO2培养箱中,根据其生长状态,按一定比例每3天传代1次。实验前将S2细胞接种在6孔板上,加入1.5ml含10%胎牛血清的Schneider培养基,待细胞生长直至密度达约80%时进行转染。
4.敲减SmD3基因:操作按脂质体Oligo-fectamineTM2000试剂说明书进行。分别用Opti-MEM培养基稀释SmD3 siRNA和转染试剂,以150nmol/L浓度进行转染。48h后行qRT-PCR观察结果以筛选出干涉效率较高的siRNA序列进行后续实验。siRNA信息描述详见表1。

表1 siRNA序列
5.过表达SmD3基因:使用Effectene Transfection Reagent试剂进行SmD3瞬时转染实验。每孔共转染0.8μg质粒,将待转染的质粒和6.4μl Enhancer混合,用buffer EC补足至100μl,振荡混匀,静置5min。然后加入20μl Effectene Transfection Reagent颠倒混匀,以形成DNA-脂质体复合物。最后,加入600μl Opti-MEM,混匀后加入细胞中,培养48h。详细的转染质粒如下:对照组为ub-Gal4+pUAS-attB;SmD3 过表达组为ub-Gal4+pUAS-attB-GFP+pUAS-attB-SmD3。
6.免疫荧光染色检测U2A蛋白的表达:将上述步骤中培养24h的S2细胞,接种于放置有圆形玻片的24孔板内,继续培养24h后进行免疫荧光实验。用4%多聚甲醛(paraformaldehyde,PFA)固定20min,用含0.1% 聚乙二醇辛基苯基醚(TritonX-100)的PBS清洗3次,每次10min,用5%的牛血清白蛋白(bovine serum albumin,BSA)封闭30min,加入兔抗U2A抗体于室温孵育1h后再用含0.1% TritonX-100的PBS清洗3遍。再用二抗在室温孵育1h,最后用含0.1% TritonX-100的PBS清洗,去除多余的二抗,用100μl 1.0mg/ml的Hoechst33342染DNA 15min后,用80%甘油封片。S2细胞免疫荧光图片通过荧光显微镜采集,每幅图像取3个不同视野,并根据像素及荧光强度利用进行荧光强度定量分析。
7.qRT-PCR检测转染后S2细胞SmD3 mRNA表达水平:将培养48h的各组S2细胞采用TRIzol法提取总RNA,紫外分光光度计测量RNA浓度。采用反转录试剂盒合成cDNA。SYBR荧光染料、cDNA、引物用来配制PCR反应体系。反应条件为95℃、2min;95℃、15s,60℃、20s,共40个循环。Ct值应用2-ΔΔCt方法处理,每个基因的相对表达量按照内参的表达量进行标准化处理。详细的引物序列见表2。
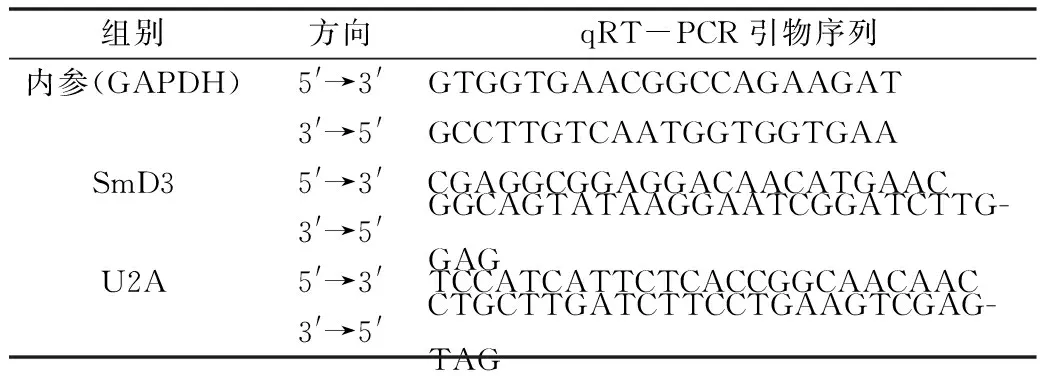
表2 qRT-PCR引物
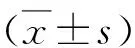
结 果
1.SmD3 siRNA干涉效率验证:在S2细胞中以150nmol/L浓度分别转染SmD3 siRNA-1、SmD3 siRNA-2干扰片段后,荧光显微镜检测可观察到FAM发出的绿色荧光信号(图1A),通过计算FAM阳性细胞比例,表明转染效率为(25.330±2.556)%(图1B)。qRT-PCR结果表明,各siRNA转染组SmD3 mRNA相对表达水平有不同程度的下调,以SmD3 siRNA-1的转染组下调最为明显,可下调约70%(图1C),因此选择SmD3 siRNA-1进行后续的实验。
2.SmD3 siRNA在果蝇S2细胞中对U2A表达水平的影响:免疫荧光结果显示(图2A),与对照组比较,SmD3 siRNA-1组的U2A蛋白荧光信号强度增强(2.177±0.197 vs 9.988±0.473),差异有统计学意义(P=0.000),表明U2A蛋白表达水平增高(图2B)。qRT-PCR结果显示,与对照组比较,SmD3 siRNA-1组的U2A mRNA相对表达水平上调约20%,差异有统计学意义(P=0.000),见图2C。

图1 SmD3 siRNA转染效率及干涉效率验证A.siRNA片段FAM荧光基团表达情况(×400),FAM(绿色),DNA(蓝色),标尺30μm;B.FAM阳性细胞所占比例; C.对照组和SmD3 siRNA组细胞中SmD3 mRNA的相对表达水平,*P=0.000
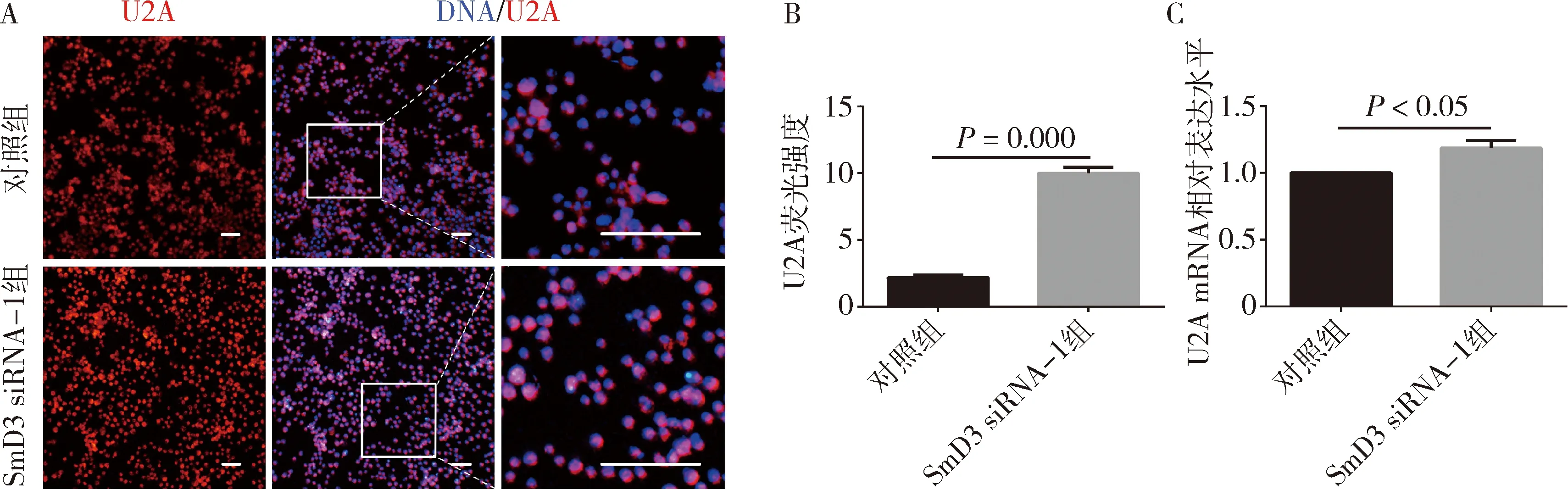
图2 敲减SmD3导致S2细胞内U2A表达水平升高A.对照组和SmD3 siRNA-1组进行免疫荧光染色(×400),U2A(红色),DNA(蓝色),标尺30μm;B.对照组和SmD3 siRNA-1组中U2A蛋白荧光信号强度柱状图;C.对照组和SmD3 siRNA-1组细胞中 U2A mRNA的相对表达水平
3.成功构建pUAS-attB-SmD3克隆并在S2细胞中实现SmD3过表达:根据SmD3 CDS序列,设计了1对含有Xhol Ⅰ和Xbal Ⅰ酶切位点的引物,进行PCR扩增。将PCR产物克隆至pUAS-attB载体中,获得重组质粒pUAS-attB-SmD3,经限制性内切酶验证和克隆片段的测序、对比,证实了克隆片段的可靠性,具体流程详见图3。在ub-Gal4驱动下,于果蝇S2细胞中同时表达pUAS-attB-GFP与pUAS-attB-SmD3。荧光显微镜检测可观察到pUAS-attB-GFP自发的绿色荧光信号(图4A),通过计算GFP阳性细胞比例,表明转染效率为(22.070±0.706)%,差异有统计学意义(P=0.000,图4B)。培养两天后进行qRT-PCR实验,图4C结果显示SmD3 mRNA相对表达水平上调至对照组20倍以上。
4.SmD3过表达在果蝇S2细胞中对U2A表达水平的影响:免疫荧光结果显示(图5A),与对照组比较,SmD3过表达组的U2A蛋白荧光信号强度减弱(2.725±0.062 vs 1.286±0.044),差异有统计学意义(P=0.000),表明U2A蛋白表达水平降低,见图5B。qRT-PCR结果显示,与对照组比较,SmD3过表达组的U2A mRNA相对表达水平下调约50%,差异有统计学意义(P=0.000),见图5C。
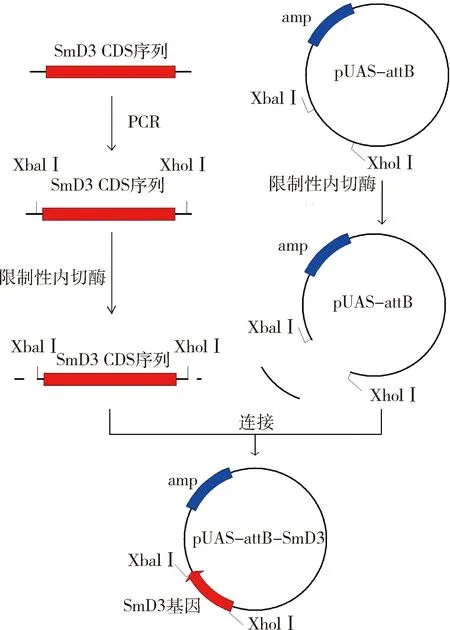
图3 构建pUAS-attB-SmD3克隆的流程
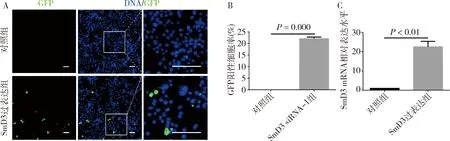
图4 SmD3基因过表达转染效率及SmD3 mRNA相对表达水平A.pUAS-attB-GFP荧光蛋白表达情况(×400),GFP(绿色),DNA(蓝色),标尺30μm;B.GFP阳性细胞所占比例;C.对照组和SmD3过表达组细胞中SmD3 mRNA的相对表达水平
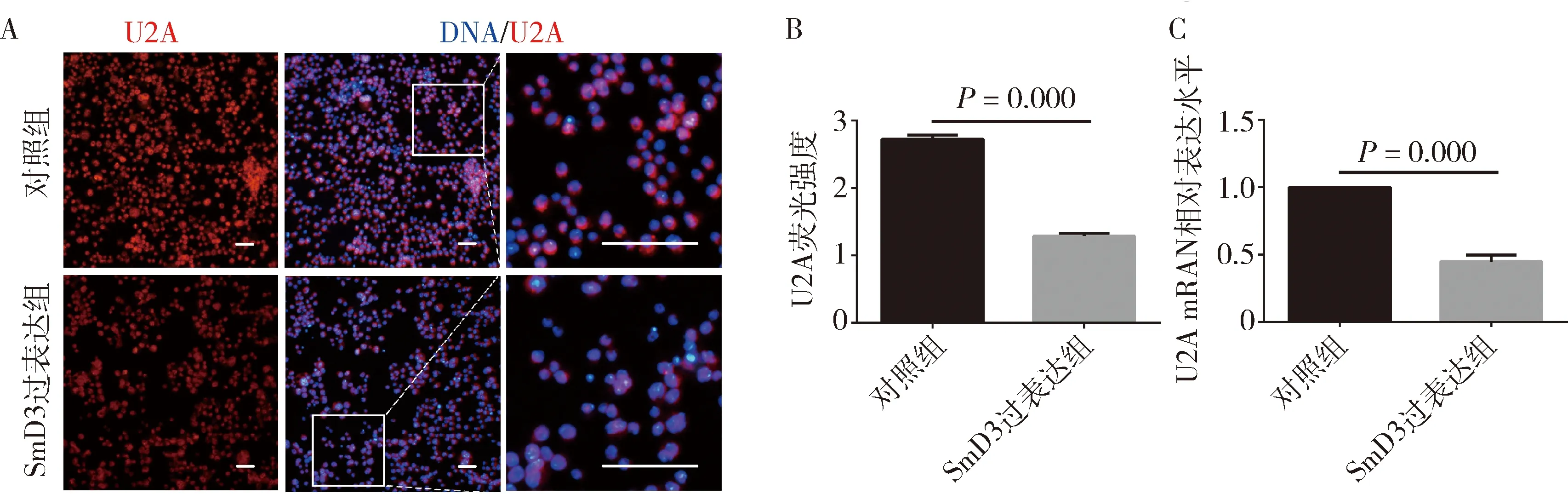
图5 过表达SmD3基因导致S2细胞内U2A表达水平下降A.对照组和SmD3过表达组进行免疫荧光染色(×400),U2A(红色),DNA(蓝色),标尺30μm;B.对照组和SmD3过表达组中U2A蛋白荧光信号强度柱状图;C.对照组和SmD3过表达细胞中U2A mRNA的相对表达水平
讨 论
剪接体是一个高度动态的分子机器,在真核生物进化中具有保守性,对于真核生物维持正常的生命活动至关重要,也是蛋白质分子多样性产生的机制之一[11,12]。pre-mRNA的剪接是真核生物基因表达的关键一步,剪接体功能异常将导致严重的后果,许多人类疾病都归咎于基因的错误剪接或剪接体的错误调控,例如地中海贫血、白血病和乳腺癌等[13,14]。
剪接体的装配过程是复杂有序的,细胞质中的运动神经元生存蛋白(survival motor neuron,SMN)复合体和经过对称二甲基化修饰的Sm/LSm蛋白结合共同形成SMN-Sm复合体,接着细胞核内小分子RNA(U snRNA),包括U1、U2、U4、U5和U6 snRNAs。通过其Sm位点保守序列(AUUUUUG)与SMN-Sm复合体结合,形成剪接体snRNPs的共同结构域,最后装配完成的snRNPs在细胞核转入受体的作用下,转运至细胞核内与pre-mRNA结合,并在其他多种蛋白因子的参与下,完成剪接过程[15,16]。
剪接体的许多组成部分已经被证明具有新功能,与果蝇生殖细胞自我更新与分化有密切的关系。SmB和SmD3是oskar信使核糖核蛋白(mRNP)的特异性组分,而oskar mRNP合适的定位是果蝇卵母细胞分化及成熟所必需的[17]。精氨酸甲基转移酶5(protein arginine methyltransferase 5,PRMT5)在剪接体装配过程中对富含精氨酸和氨基乙酸残基的序列区的Sm蛋白(SmD3、SmB/B′、LSm4)进行对称二甲基化修饰,PRMT5缺失将导致果蝇卵子发生过程受阻[18]。剪接体因子Prp22、Prp38通过影响卵子发生早期支持细胞中染色质的扩散,导致染色体形态受损,影响卵子发生过程[19]。而剪接体成分U2A则已被证实具有调控果蝇精原细胞分化的功能[8]。
果蝇S2细胞是优于哺乳动物细胞的表达外源蛋白的细胞模型,当带有目的基因的载体通过实验操作整合到细胞基因组中,能够完成正确的转录、翻译及蛋白加工过程,所表达的目的蛋白质在结构、功能上与天然的基本相同[20]。同时,果蝇S2细胞是半悬浮细胞,具有易于培养、生长速率较快等优势。因此,本研究利用果蝇S2细胞为体外模型,通过改变SmD3的表达后检测U2A的表达水平,探究两者的相互作用,借助功能已知的U2A来阐述SmD3可能的新功能。通过研究发现,当SmD3基因表达水平下降时,U2A的表达水平补偿性上调。有趣的是,当SmD3基因表达水平上调时,一定程度上抑制了U2A的表达。由此,笔者提出了一个工作模型,果蝇S2细胞中SmD3与U2A处于相互竞争的关系。因此,笔者提出猜想,SmD3作为被鉴定到的参与调控生殖干细胞自我更新及分化的因子之一,可能通过影响U2A发挥作用。本研究仅在体外实验中通过S2细胞初步提出SmD3对U2A的影响,而SmD3对果蝇生殖干细胞的调控机制还需进行体内实验来进一步探索。本研究中通过干涉效率验证筛选到SmD3 siRNA-1片段,可为后续研究中制备SmD3 RNAi果蝇提供靶向序列。此外,本研究成功构建了pUAS-attB-SmD3克隆,可为后续研究中SmD3转基因果蝇的制备提供过表达质粒,以便进行果蝇体内实验进一步验证及深入研究。
综上所述,本研究结果证实果蝇S2细胞中SmD3与剪接因子U2A发挥相互拮抗的作用,从而可以为进一步研究剪接体核心蛋白SmD3的生物学功能提供思路。
猜你喜欢
广东药科大学学报(2022年3期)2023-01-04
生物学通报(2022年1期)2022-11-22
学苑创造·A版(2022年3期)2022-03-29
烟台果树(2021年2期)2021-07-21
江西农业学报(2021年4期)2021-04-20
学苑创造·A版(2019年6期)2019-07-11
福建基础教育研究(2019年2期)2019-05-28
江西医药(2018年8期)2018-10-24
罕少疾病杂志(2017年2期)2017-02-23
西南医科大学学报(2015年1期)2015-08-22
