
耐力运动对GK大鼠肝脏内质网应激和胰岛素信号通路的影响
2019-10-11 09:03戈哲孙易漆正堂丁树哲
中国运动医学杂志 2019年9期
戈哲 孙易 漆正堂 丁树哲
1华东师范大学青少年健康评价与运动干预教育部重点实验室(上海200241)
2华东师范大学体育与健康学院(上海200241)
内质网应激是由于内质网环境发生变化,例如缺氧、化学毒物等多种因素抑制内质网蛋白质加工的糖基化或者导致蛋白质二硫键的错配,从而导致未折叠蛋白在内质网上的积累从而引发下游一系列信号通路的过程。1988年,Kozutsumi等提出了内质网应激存在信号转导的过程[1]。内质网应激所引发的未折叠蛋白反应(unfolded protein response,UPR)信号通路通过各种途径对胰岛素信号通路存在一定的影响,例如肌醇需求激酶1(inositol-requiring enzyme-1,IRE1)的激活导致下游c-Jun氨基末端激酶(c-Jun N-terminal kinase,JNK)磷酸化,JNK磷酸化再导致下游胰岛素受体底物1(insulin receptor substrate 1,IRS-1)异位点的磷酸化,进而诱发胰岛素抵抗。体外实验表明,ER应激可引起肝脏、肌肉和脂肪等外周组织的胰岛素抵抗[2]。IRS-1的正常磷酸化位点是酪氨酸,IRS-1特定的丝氨酸位点磷酸化则会抑制胰岛素信号通路[3]。肝脏是人体胰岛素作用的主要器官之一,在正常生理状态下,胰岛素通过与肝脏胰岛素受体结合从而摄取血液中的葡萄糖进而加强肝糖原的合成来维持体内糖代谢的稳定。然而,在胰岛素抵抗的状态下,肝脏摄取葡萄糖的能力下降[4]。Ozcan等[5]利用衣霉素诱导肝细胞ER应激,发现胰岛素刺激的Akt磷酸化和IRS-1酪氨酸磷酸化受到抑制,同时IRS-1丝氨酸磷酸化增强。有研究表明内质网应激增强和JNK通路的激活干扰了胰岛素信号通路,造成了胰岛素抵抗[6]。Jiro等[7]研究发现与正常小鼠对比,JNK1敲除小鼠肝脏中IRS-1酪氨酸磷酸化增强,胰岛素受体功能恢复。这也提示了JNK1是诱发IRS-1异位点磷酸化的关键。IRE1-JNK通路是内质网应激引发胰岛素抵抗的经典信号通路,而目前众多研究表明长期的耐力运动能够缓解2型糖尿病的症状,降低体内血糖水平,提高胰岛素受体敏感性。为探索运动是否通过内质网应激信号通路来影响2型糖尿病的发展,本研究以GK大鼠为2型糖尿病模型,联合运动干预,从内质网应激IRE1-JNK信号通路的视角探索肝脏胰岛素抵抗的机制,以期为2型糖尿病的预防和治疗提供理论依据。
1 材料与方法
1.1 实验动物
清洁级雄性12周龄GK大鼠(Goto Kakiski Wistar rats,自发2型糖尿病动物模型)和Wistar大鼠,均由上海斯莱克实验动物有限公司提供。实验动物生产许可证号:SCXK(沪)2012-0002。实验动物使用许可证号:SCXK(沪)2012-0002。动物饲料喂养,实验大鼠自由饮食、饮水,1~2天更换一次垫料。大鼠饲养环境温度为20℃~24℃,相对湿度维持在50%~70%,自然光照,正常室内通风。
1.2 GKGK大鼠糖尿病模型的确定
对购买的大鼠立即进行空腹血糖测定,确定GK大鼠糖尿病模型的建立成功。见表1。

表1 Wistar大鼠与GK大鼠运动干预前空腹血糖的测定(mmol/L)
1.3 实验动物的分组与运动方案
GK 大鼠16只,随机分为糖尿病组(D组)和糖尿病运动组(DE组),每组8只。对照Wistar雄性大鼠16只,随机分为正常组(C组)和正常运动组(E组),每组8只。DE及E组进行为期6周的跑台耐力运动,速度按周递增分别为15、16、17、18、19、20 m/min,持续时间为1 h,每周运动6天,周日休息。运动过程中不定期称重。
1.4 腹腔注射葡萄糖耐量试验
6周耐力训练后进行腹腔注射葡萄糖耐量试验(intraperitoneal glucose tolerance trial,IPGTT),大鼠禁食12小时后称重,计算所需葡萄糖量,腹腔注射1.0 g葡萄糖/kg,采用剪尾法取尾静脉血,检测0 min(空腹)、15 min、30 min、60 min、90 min,120 min大鼠全血血糖水平。
1.5 实验动物的处死与取材
末次运动结束,实验大鼠空腹24小时后处死。取肝脏,迅速装入已经标记好的冻存管中,立即置于液氮中速冻,之后转入-80℃冰箱保存,指标待测。
1.6 实时荧光定量PCRPCR
冰上称取30 mg肝脏组织,然后采用Trizol法提取肝脏中RNA,按照Trizol试剂说明书进行提取。提取后采用超微量分光光度计NanoDrop测定提取RNA的浓度和纯度。本实验使用TOYOBO的逆转录试剂盒进行RNA的反转录。采用ABI Step One型实时荧光定量PCR仪器检测Bip、IRE1、JNK1、AKT2、JNK2、IRS1及βactin的mRNA相对表达量,β-actin为内参。实验引物均由上海生物工程有限公司合成,具体引物序列如下(表2),PCR结果数据分析采用2—ΔΔCT法进行分析。

表2 基因引物序列
1.7 Western Blotting
本实验蛋白提取主要采用试剂为RIPA细胞裂解液(Thermo Scientific)和蛋白酶抑制剂(Roche),按罗氏蛋白酶抑制剂说明书,每10 ml RIPA裂解液中加入1片蛋白酶抑制剂和1片磷酸酶抑制剂。取组织40 mg置于放有磁珠的研磨管中,加入400 μl的裂解液(RIPA Lysis Buffer+1片蛋白酶磷酸酶抑制剂)。OMINI:OM-RP24型磁珠均质匀浆仪匀浆。每匀浆1次,置于冰上5 min。共匀浆3次,最后1次置于冰上20 min。然后4度离心,14000 g,20 min。取上清,BCA蛋白浓度测定调平蛋白液浓度后进行SDS-PAGE实验。实验结果采用Alpha:FluorChem HD2凝胶成像系统进行拍照分析。其中 Bip、JNK1、JNK2、phospho-JNK(phospho T183+T183+T221)抗体均购于Abcam公司,AKT抗体购于CST公司。
1.8 统计方法
本实验各数据结果均采用±s的方式表示,统计软件为EXCEL、SPSS。采用独立样本t检验和双因素方差分析统计实验数据,两两比较采用LSD法。P<0.05为差异有统计学意义。
2 结果
2.1 耐力运动对大鼠体重的影响及耐力运动后血糖IPIP--GTTGTT测试
与C、E组相比,D、DE组大鼠体重均表现为低体重,E组体重相较C组呈下降的趋势。DE组相较D组,体重同样呈现下降趋势。6周耐力训练后血糖IPGTT测试结果表明,D、DE组大鼠IPGTT测试中血糖整体水平明显高于C、E组大鼠(P<0.001),并且其血糖水平在注射葡萄糖之后15 min后快速升高,且在60 min时,D组血糖水平达到峰值,DE组则已经越过峰值开始下降,并且DE组血糖自60 min后显著低于D组(P<0.01),C组和E组15 min后血糖水平达到峰值。见图1。

图1 运动对大鼠体重的影响及6周耐力运动后大鼠血糖IPGTT测试结果
2.2 耐力运动对大鼠BipBip、JNKJNK等内质网应激和胰岛素信号通路相关mRNAmRNA水平的影响
与C组相比,E组的Bip mRNA水平无显著性差异,D组的Bip mRNA有上升趋势(P=0.06)。与E组相比,DE组的JNK1 mRNA 的表达有降低趋势(P=0.06),而其余基因的mRNA水平均无明显差异。见图2。
2.3 耐力运动对BipBip、JNKJNK等内质网应激和胰岛素信号通路相关蛋白水平的影响
如图3所示,与C组相比,D组JNK1蛋白表达升高(P<0.05)。与E组相比,DE 组的JNK2蛋白表达降低(P<0.01)。与C组相比,D组AKT蛋白表达下降(P<0.01);与E 组相比,DE组的AKT蛋白表达下降(P<0.01)。

图2 运动对大鼠肝脏Bip、IRE1、JNK1、JNK2、AKT2、IRS1基因表达水平的影响
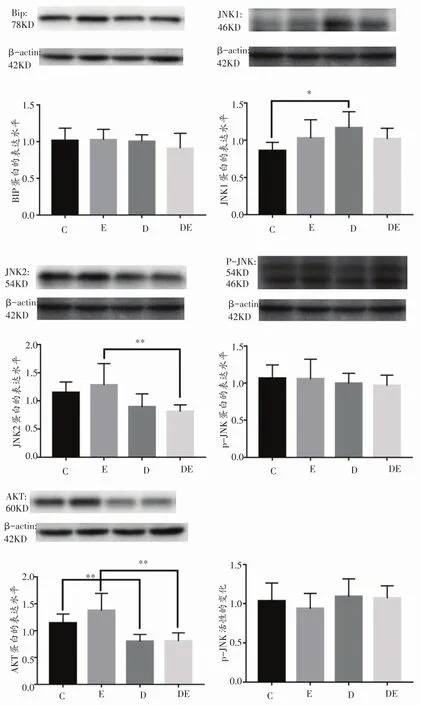
图3 运动对大鼠肝脏Bip、JNK1、JNK2、p-JNK、AKT蛋白表达水平的影响
3 分析和讨论
3.1 耐力运动对大鼠体重的影响及耐力运动后血糖IPIP--GTTGTT测试
有研究表明GK大鼠由于2型糖尿病导致体重增速减缓,发病后体重显著低于Wistar大鼠[8],本研究GK大鼠体重符合实验预期。而长期耐力训后大鼠表现出体重降低的趋势。2型糖尿病的主要病理生理特征是高血糖结合胰岛素抵抗。本研究结果表明,与C、E组相比,D、DE组的降血糖能力明显下降。C组、E组IPGTT测试结果无明显差异,注射葡萄糖溶液60 min~120 min后,DE组血糖水平显著低于D组,提示6周的跑台耐力运动可以改善GK大鼠血糖水平,有利于维持血糖水平稳态。
3.2 耐力运动对大鼠内质网应激与胰岛素抵抗相关分子表达的影响
静息状态下,免疫球蛋白重链结合蛋白(immunoglobulin heavy chain-binding protein,Bip)与 PERK、IRE1、ATF6处于结合状态。随着内质网应激的发生,三者从Bip上释放出来,进而激活下游信号通路,引发UPR。Bip是内质网应激的标志物,在内质网应激的情况下,Bip表达升高[9]。Ozcan等[5]研究表明Bip表达增加后,通过IRE1-JNK通路使IRS1丝氨酸异位点磷酸化增加,最终导致胰岛素抵抗。然而,Jennifer研究表明,用牛磺熊去氧胆酸、四苯基乙酸和过表达Bip来减缓内质网应激,肌细胞在棕榈酸盐的诱导作用下并未出现胰岛素抵抗减缓的现象。这也表明内质网应激并不是导致胰岛素抵抗的唯一因素[10]。最近研究表明脂肪组织巨噬细胞分泌的外泌体上所携带的micro RNA155可以直接抑制胰岛素信号通路[11],提示导致胰岛素抵抗的原因可能是复杂多样的。内质网应激的发生会导致IRE1的激活,从而引发进一步UPR信号反应。而我们的研究结果发现,相对正常组,D组大鼠Bip蛋白表达并未升高,仅Bip mRNA水平有上升趋势(P=0.06),而IRE1同样无显著性差异,表明本实验GK大鼠内质网并未发生应激。这也说明了虽然大量证据表明内质网应激会导致胰岛素抵抗,但导致胰岛素抵抗的原因可能并非只有内质网应激,可能还存在其它的因素。并且,众多研究表明长期运动能够减缓内质网应激,降低胰岛素抵抗。然而,本研究结果显示运动并未减缓内质网应激,推测6周的跑台耐力训练的运动量可能不足以降低Bip和IRE1的表达。
JNK是MAPK家族中的重要一员,包括JNK1、JNK2、JNK3。其中JNK1和JNK2在功能上存在着较大的差别。Ruo等[12]用高果糖膳食喂养小鼠,发现肝脏中IRE1表达增加,JNK磷酸化显著上升,IRS1丝氨酸位点磷酸化显著增强,并且伴随着AKT磷酸化的下降,这表明内质网应激可以通过激活JNK来诱发胰岛素抵抗。Jiro等[7]研究发现,肥胖小鼠中伴随着JNK活性的异常升高。敲除JNK1基因,肥胖症状则有所缓解,胰岛素抵抗的症状有明显的改善。他们还发现高脂膳食能够诱导小鼠肌肉、肝脏及脂肪组织JNK1的激活,这表明脂肪可能是导致JNK1激活的关键因素。Sharma等[13]通过siRNA敲低IRE1,发现饱和脂肪酸能在肝细胞中直接诱导JNK的激活,这表明IRE1的激活并不是JNK激活的主要因素,脂肪在肝细胞中的堆积也能导致JNK激活。本研究表明,C组与D组在JNK1 mRNA水平没有显著性差异。与E组相比,DE组JNK1 mRNA水平有降低的趋势。推测运动和糖尿病二者共同作用调节,导致DE组JNK1转录降低,但其具体机制仍不明确。与C组相比,D组的JNK1蛋白表达明显升高,且实验结果显示JNK1磷酸化水平并未出现显著性差异,这提示JNK1是通过总蛋白增加来引发胰岛素抵抗,但本研究中JNK1的激活因素究竟是什么,有待进一步研究。从JNK2 mRNA水平来看,各组之间无显著性差异,而从蛋白水平来看,与C组相比,D组JNK2蛋白有下降趋势,并且相对E组而言,DE组JNK2蛋白水平显著下降,可能是糖尿病大鼠JNK2 mRNA翻译过程受阻,或者其转录后受到细胞内非编码RNA的调控,其具体机制尚不明确。Gurol等[14]研究发现JNK2基因敲除的动物并未表现出任何胰岛素敏感性和体重的改变。由此,我们不难得出,JNK1和JNK2两种同源蛋白在细胞内共同调节生理功能时,JNK2对胰岛素信号通路似乎没有明确直接的影响,而JNK1则与胰岛素信号通路存在着密切的关系。
IRS-1是胰岛素信号通路中的关键受体蛋白,本研究表明IRS1 mRNA水平各组之间均无显著性差异,这表明JNK1导致的胰岛素敏感性降低与IRS1上游信号的变化无关,而是直接影响IRS1磷酸化以及下游分子的变化。丝氨酸/苏氨酸蛋白激酶AKT又被称为蛋白激酶B(protein kinase B,PKB),有几种亚型,如AKT1、AKT2,而AKT2与胰岛素信号通路的关系更为密切。Frankish研究表明,AKT2基因敲除的小鼠表现出2型糖尿病的症状[15]。研究表明,2型糖尿病中AKT的表达被抑制,例如Christ发现2型糖尿病病人肌细胞AKT表达降低[16],王亚等研究发现2型糖尿病大鼠肝组织AKT表达降低[17],这表明胰岛素信号通路处于抑制状态。本研究结果表明,AKT2 mRNA水平上各组之间无显著性差异,但从C组和D组蛋白水平来看,D组AKT蛋白水平低于C 组(P<0.01)。而且,DE 组AKT蛋白表达水平低于E 组(P<0.01)。我们推测,由于D组和DE组同是2型糖尿病,存在着严重的胰岛素抵抗,所以其胰岛素信号通路被抑制,AKT表达下降。然而从C组与E组以及D组与DE组比较来看,其AKT蛋白表达相互之间不存在显著性差异,推测6周的跑台运动的运动量不足以改善胰岛素信号通路。
4 结论
本研究显示肝脏组织中,相较于正常大鼠,2型糖尿病大鼠肝脏JNK1蛋白表达水平显著升高,其JNK2水平无明显差异,表明胰岛素抵抗的发生与JNK1的表达水平的升高密切相关。此外,6周跑台耐力运动并未影响大鼠肝脏内质网应激水平和GK大鼠肝脏胰岛素信号通路,但其能改善GK大鼠血糖水平,有利于维持血糖水平稳态。
猜你喜欢
波谱学杂志(2022年1期)2022-03-15
昆明医科大学学报(2022年1期)2022-02-28
临床肝胆病杂志(2021年7期)2021-12-26
现代临床医学(2021年1期)2021-01-26
中国体育教练员(2018年2期)2018-07-23
车迷(2017年10期)2018-01-18
分析化学(2017年12期)2017-12-25
安徽医科大学学报(2015年9期)2015-12-16
癌变·畸变·突变(2015年3期)2015-02-27
癌变·畸变·突变(2015年3期)2015-02-27
