
利用基因工程改造的大肠杆菌合成蒎烯
2019-09-27 02:26王诗语王鹤蓉黄子豪陈飞张霞赵彦斌朱晨刘刚陈惠鹏
生物技术通讯 2019年4期
王诗语,王鹤蓉,黄子豪,陈飞,张霞,赵彦斌,朱晨,刘刚,陈惠鹏
1. 安徽大学 物质科学与信息技术研究院,安徽 合肥230601;2. 军事科学院 军事医学研究院,北京100850;3. 吉林大学 生命科学学院,吉林 长春130012
萜类代谢途径中,中间产物异戊二烯焦磷酸(isopentenyl pyrophosphate,IPP)和二甲基烯丙基焦磷酸(dimethylallyl diphosphate,DMAPP)是萜类化合物生物合成的必需前体物质。1 分子DMAPP与不同分子的IPP 在异戊烯转移酶的作用下生成不同的萜类前体[15],这些前体在各种萜类合酶(terpenesynthases,TPS)催化下合成不同种类的萜类化合物[16]。自然界中,有机体合成DMAPP 和IPP 有2 种代谢途径,即1-脱氧木酮糖-5-磷酸(1-deoxy-D-xylulose-5-phosphate,DXP)途径和甲羟戊酸(mevalonate,MVA)代谢途径。DXP 途径又被称为甲基赤藓糖醇磷酸(MEP)途径,存在于部分古细菌、动物、大多数藻类及高等植物的叶绿体中,它的前体物质与MVA 代谢途径的前体物质有所不同,是丙酮酸和三磷酸甘油醛。MVA 代谢途径普遍存在于真核细胞中,在部分古细胞和革兰阳性细菌中也存在[17]。MVA 代谢途径前体物质是乙酰辅酶A,整个过程有6 种蛋白酶参与反应,分别为mvaE表达的蛋白[具有乙酰辅酶A 乙酰 转移酶(acetyl-CoA acetyltransferase)和3-羟基-3-甲基戊二酸辅酶A 还原酶(3-hydroxy-3-methyl glutaryl coenzyme A reductase,HMGR)2 种活性功能]、mvaS表达的3-羟基-3-甲基戊二酰辅酶A 合成酶(3-hydroxy-3-methyl glutaryl coenzyme A synthase,HMGS)、ERG12表达的甲羟戊酸激酶(mevalonate kinase,MK)、ERG8表达的磷酸甲羟戊酸激酶(phosphomevalonate kinase,MPK)、ERG19表达的甲羟戊酸焦磷酸脱羧酶(mevalonatepyrophosphate decarboxylase,MPD)、IDI表达的异戊烯焦磷酸异构酶(isopentenyl diphosphate isomerase,IDI)。其中IDI 可催化IPP 和DMAPP 的相互转化,在MVA 代谢途径中起关键作用[18]。
大肠杆菌具有遗传背景清晰、生长代谢快、易于人工改造等优点,一直是生物制造应用领域最为热门的底盘细胞之一。在利用大肠杆菌合成外源萜类化合物的过程中,大肠杆菌可以通过自身DXP 代谢途径合成萜类前体物质IPP 和DMAPP。但其自身代谢产生的IPP 和DMAPP 并不能满足萜类化合物的生产需求,有必要对大肠杆菌进行改造,修饰自身的或引入异源的IPP 和DMAPP 合成途径[19]。Meng 等利用流平衡分析(flux balance analysis,FBA)对微生物通过MVA途径和DXP 途径合成IPP 做了相关模拟,结果表明大肠杆菌中能够共存这2 种代谢途径,并且这2 种途径共存时能提高IPP 的理论最大产值[20]。
随着单萜化合物蒎烯逐渐获重视,以大肠杆菌为底盘生物构建“微生物工厂”合成蒎烯已被证明是一种可持续获取蒎烯的新途径。
通过调研,本研究选取了高效GPPS、PS基因以及高产IPP 和DMAPP 的MVA 代谢途径,对来源于北美巨冷杉的GPPS和PS基因序列经密码子优化后人工合成,分别构建共表达载体和融合表达载体,与来自酵母和粪肠球菌异源杂合高效MVA代谢途径共同转化大肠杆菌BL21(DE3)后诱导表达,其中MVA 上游途径为来源于粪肠球菌的mvaE和mvaS基因簇,MVA 下游途径为来源于酿酒酵母菌的ERG12、ERG8、ERG19和IDI基因,将这4 个基因分别利用传统的多顺子模型和Bio-Brick 方法进行拼接后构建2 种不同方案的MVA代谢途径。将融合表达载体和MVA 代谢途径共转化大肠杆菌,高效表达蒎烯,为微生物工业化生产蒎烯奠定初步基础。
1 材料与方法
1.1 材料
大肠杆菌DH5α感受态细胞购自康为世纪生物科技有限公司;大肠杆菌BL21(DE3)感受态细胞购自天根生化科技(北京)有限公司;pET-24a(+)与pET-30a 为本实验室保存;pETDuet-1 购自Novagen 公司;pMD18-T 购自宝生物工程有限公司;pACYCDuet-a 购自BioVector 公司;限制性内切酶、T4DNA 连接酶、Q5 超保真2×Master Mix、质粒提取试剂盒、胶回收试剂盒、清洁回收试剂盒、DNA 快速连接试剂盒、SOC 培养基均购自NEB 公司;酵母基因组DNA 提取试剂盒和溶细胞酶购自天根生化科技(北京)有限公司;α蒎烯和β蒎烯标准品购自Sigma 公司;抗生素购自生工生物工程(上海)股份有限公司。
高密度培养基:葡萄糖10 g,酵母提取物11.5 g,胰蛋白胨19.5 g,硫酸铵4 g,三水磷酸氢二钾18 g,磷酸二氢钾3 g,蒸馏水定容至1 L,115℃高压灭菌20 min;硫酸镁1.2 g,微量元素母液250 μL 用0.22 μm 膜过滤除菌后加入培养基中。微量元素母液:七水硫酸亚铁2.8 g,四水氯化锰2 g,七水硫酸钴2.8 g,二水氯化钙1.5 g,二水氯化铜0.2 g,七水硫酸锌0.3 g,溶于250 mL 1 mol/L 盐酸溶液中,用0.22 μm 膜过滤除菌。
1.2 GPPS-PS表达载体的构建
1.2.1 目的基因的获取 从GenBank 中获取北美巨冷杉GPPS和PS基因序列,用DNAssist 软件分析基因序列详情,根据密码子简并性及大肠杆菌密码子偏爱性对基因序列进行优化,改造稀有密码子并去掉常用的限制性酶切位点,设计好的核酸序列由北京奥科鼎盛生物科技有限公司人工合成,分别命名为pGPPS-0 和pPS-0。
幼儿美术活动多种多样,而作为一名幼儿教师,在立足于传统教学的同时,应该不断提高自身能力和专业水平,探索创新,寻求更多更好的方法,为幼儿身心全面发展提供机会。绘本作为幼儿绘画活动中一种较好的媒介,以其独特的表达系统契合幼儿的心理特点,有利于幼儿从中得到积极的情感体验,从而激发幼儿将内心感受绘于纸上的愿望。

表1 引物序列(1)
1.2.2 构建共表达载体 共表达GPPS和PS基因分别由2 个启动子控制并独立表达成2 个蛋白,以含目的基因的质粒为模板设计引物(表1),扩增目的基因片段,并在GPPS基因两端引入BamHⅠ和HindⅢ酶切位点、PS基因两端引入NdeⅠ和BglⅡ酶切位点,进行PCR 扩增[98℃ 30 s;98℃20 s,58℃(GPPS)/59℃(PS)20 s,72℃40 s(GPPS)/60 s(PS),30 个循环;72℃ 2 min],将扩增的GPPS和PS基因分别酶切连入不同的多克隆位点,构建pETDuet1-GPPS-PS,鉴定正确的质粒交由华大公司测序。
1.2.3 构建融合表达载体 首先将GPPS去掉终止密码子后利用一个柔性的linker 串联PS基因,再以pET-24a(a)为基础构建融合表达载体。根据融合PCR 的原理设计扩增GPPS和PS的2 对引物(表2),在GPPS的5'端引入BamHⅠ酶切位点,PS的3'端引入XhoⅠ酶切位点,基因间linker 序列为GGTTCTGGT。第1 次PCR 分别扩增GPPS和PS基 因[98℃ 30 s;98℃ 20 s,62℃(GPPS)/63℃(PS)20 s,72℃ 50 s(GPPS)/60 s(PS),30 个 循环;72℃ 2 min];第2次扩增以第1次扩增的GPPS和PS的PCR 胶回收产物为模板、GPPS2-F与PS2-R 为引物,扩增GPPS-PS融合片段(98℃30 s;98℃ 20 s,63℃ 20 s,72℃ 2 min,30 个循环;72℃2 min)。对扩增后的胶回收产物和载体pET-24a(+)进行酶切连接,产物转化大肠杆菌感受态细胞,转化子酶切测序鉴定,鉴定正确的质粒命名为pET24a-GPPS-PS。
1.2.4 构建共表达及融合表达工程菌 分别将共表达载体pETDuet1-GPPS-PS 和融合表达载体pET24a-GPPS-PS 转化大肠杆菌BL21(DE3),工程菌分别命名为E.hzh01 和E.hzh02。将过夜培养的E.hzh.01 和E.hzh.02 按1∶100 的比例分别接种到卡那霉素(Kan)抗性的LB 培养基中,37℃、200 r/min培养至菌液D600nm值为0.8~1.0,加入终浓度为1 mmol/L 的IPTG,30℃、200 r/min 诱导培养10 h,取全菌进行10% SDS-PAGE,分析基因表达情况。
1.2.5 蒎烯含量检测 将过夜培养的E.hzh.01 和E.hzh.02 按1∶100 的比例分别接种至含相应抗生素的100 mL 高密度培养基中,37℃、200 r/min 培养至菌液D600nm值为0.8~1.0,加入终浓度为1 mmol/L 的IPTG,在培养基上覆盖20%的正十二烷溶液,继续在30℃、200 r/min 培养72 h,利用GCMS 分别检测十二烷层和水层蒎烯含量。采用美国安捷伦公司的5975C-7890A 单四极杆气相色谱质谱联用仪和HP-5MS(60 m×0.32 mm×0.5 μm)色谱柱。质谱方法,采用选择离子扫描模式(SIM),离子源温度230℃,四极杆温度150℃。气相方法,进样口温度200℃,流速2.5 mL/min 恒流模式。分流模式,分流比50∶1;柱温箱,50℃起始,保持3 min,10℃/min 升至130℃,保持1 min,30℃/min 升至280℃,保持2 min;进样量1 μL。

表2 引物序列(2)

表3 引物序列(3)
1.3 含MVA途径产蒎烯工程菌构建
1.3.1 构建MVA 上游表达载体 设计引物(表3)扩增mvaE和mvaS基因,在其两端分别加入NcoⅠ和BamHⅠ酶切位点,以军事医学研究院生物工程研究所方宏清副研究员惠赠的mvaE和mvaS基因为模板进行PCR 扩增(98℃ 30 s;98℃ 20 s,57℃20 s,72℃2 min,30 个循环;72℃2 min)。
以pACYCDuet-1 为基础载体,通过对基础载体和目的基因PCR 产物进行酶切连接构建MVA上游途径表达载体pACYCDuet-mvaE/S。连接产物转化大肠杆菌DH5α感受态细胞,将酶切鉴定且测序正确的质粒转化大肠杆菌BL21(DE3),SDS-PAGE 验证基因mvaE和mvaS的表达。为了外源杂合MVA 代谢途径与GPPS-PS融合表达基因更加方便地转化大肠杆菌,利用PCR 方法在GPPS-PS融合基因两端分别引入BglⅡ和PvuⅠ酶切位点进行酶切连接,整合进载体pACYCDuetmvaE/S,构建成功的质粒命名为pACYCDuetmvaE/S-GPPS-PS。
1.3.2 多顺反子模型原理构建MVA 下游表达载体 提取酿酒酵母基因组作为模板,分别PCR 扩增目的基因ERG12、ERG8、ERG19和IDI,选用NEBuilder HiFi DNA Assembly Cloning Kit,根据多顺反子模型原理构建基因四串联共表达载体。利用NEB 在线工具(NEBuilder Assembly Tool)设计引物(表4)分别扩增目的片段[98℃ 30 s;98℃ 20 s,57℃(ERG12)/57℃(ERG18)/55℃(ERG19)/54.5℃(IDI)/56℃(pET-24a) 20 s,72℃45 s(ERG12、ERG8、ERG19、IDI)/180 s(pET-24a),30 个循环;72℃2 min]。
利用目的片段之间添加的同源重组片段进行重组连接,将扩增得到的目的片段和pET-24a(+)表达载体与DNA Assembly Master Mix 按照一定比例混合后,50℃温育60 min,重组反应液直接转化大肠杆菌,经酶切测序鉴定,构建成功的载体命名为pET24a-ERG12-8-19-IDI。

表4 引物序列(4)

表5 引物序列(5)
1.3.3 BioBrick 原理构建MVA 下游表达载体 以酵母基因组为模板,设计引物(表5)分别克隆目的基因ERG12、ERG8、ERG19和IDI,引物设计时分别在基因两端引入相应的酶切位点(ERG12和IDI酶切位点为NdeⅠ/XhoⅠ,ERG8和ERG19为BamHⅠ/XhoⅠ)。PCR 条件:98℃ 30 s;98℃ 20 s,61℃(ERG12)/59℃(ERG18)/59℃(ERG19)/55℃(IDI)20 s,72℃ 50 s,30 个 循环;72℃ 2 min。目的产物与pET-30a 载体分别酶切连接后,转化大肠杆菌DH5α感受态细胞,酶切测序鉴定正确的质粒分别命名pET30a-ERG12、pET30a-ERG8、pET30a-ERG19、pET30a-IDI。
进一步利用XbaⅠ和SpeⅠ互为同尾酶的原理将4 个BioBrick 单表达载体串联一起,构建4 个基因共表达载体pET30a-ERG12-8-19-IDI。以pET30a-ERG12、pET30a-ERG8 串联为例,对2 个载体分别进行双酶切,胶回收pET30a-ERG12 大片段及pET30a-ERG8 小片段后进行连接,酶切测序鉴定多基因表达载体,最后鉴定正确的质粒命名为pET30a-ERG12-8-19-IDI。
1.3.4 含MVA 代谢途径产蒎烯工程菌的构建 将pACYCDuet- mvaE/S- GPPS- PS 载体与pET24a-ERG12-8-19-IDI 或pET30a-ERG12-8-19-IDI 载体共同转化大肠杆菌,构建含MVA 代谢途径产蒎烯的工程菌E.hzh03 和E.hzh05。将工程菌E.hzh03和E.hzh05 分别接种至含相应抗生素的100 mL 高密度培养基中,37℃、200 r/min 培养至菌液D600nm值为0.8~1.0,加入终浓度为1 mmol/L 的IPTG,在培养基上覆盖20%的正十二烷溶液,继续在30℃、200 r/min 培养72 h,利用GC-MS 检测蒎烯产量。
2 结果
2.1 GPPS-PS表达载体的鉴定
分别克隆GPPS和PS基因序列,在GPPS基因两端引入BamHⅠ和HindⅢ酶切位点,PS基因两端引入NdeⅠ和BglⅡ酶切位点,酶连基因片段到表达载体pETDuet-1 中,酶切并测序显示成功构建共表达载体pETDuet1-GPPS-PS(图1A)。将共表达载体转化大肠杆菌BL21(DE3)感受态细胞构建工程菌株E.hzh.01,诱导表达后SDS-PAGE 分析表明目的条带与预期大小一致,目的基因在大肠杆菌中成功表达(图1B)。
利用融合PCR 构建GPPS和PS融合表达载体,酶切并测序显示成功构建了融合表达载体pET24a-GPPS-PS(图1C)。将融合表达载体转化大肠杆菌BL21(DE3)感受态细胞构建工程菌株E.hzh.02,诱导表达后SDS-PAGE 分析表明融合蛋白条带相对分子质量约116×103,与预期相符(图1D),融合基因在大肠杆菌中成功表达。
2.2 蒎烯含量检测方法的建立
利用GC-MS 方法检测蒎烯产量,根据蒎烯标准品的质谱分析图(图2A)确定α蒎烯及β蒎烯的定性离子和定量离子。根据蒎烯标准品的气相色谱图(图2B)确定α蒎烯及β蒎烯的保留时间。分别以α蒎烯及β蒎烯的浓度与色谱峰面积制作α蒎烯及β蒎烯的浓度标准曲线(图2C),结果显示α 蒎烯和β 蒎烯的RSD1111 值分别为2.27%和2.12%,证明GC-MS 检测蒎烯产量的方法重现性良好。
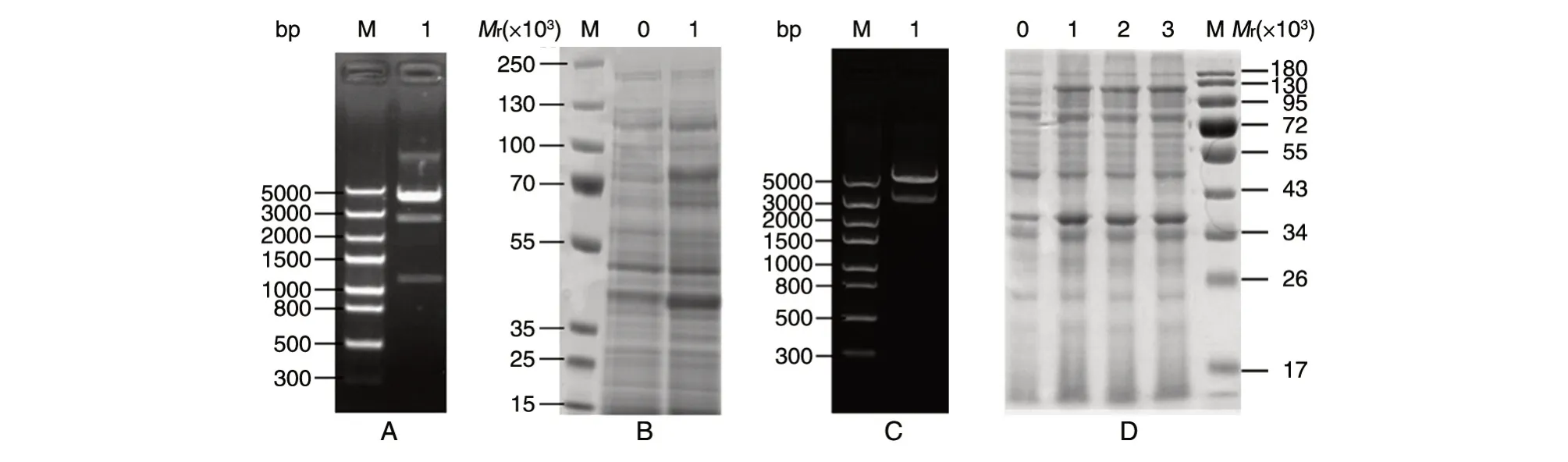
图1 表达载体酶切鉴定和诱导表达(1)
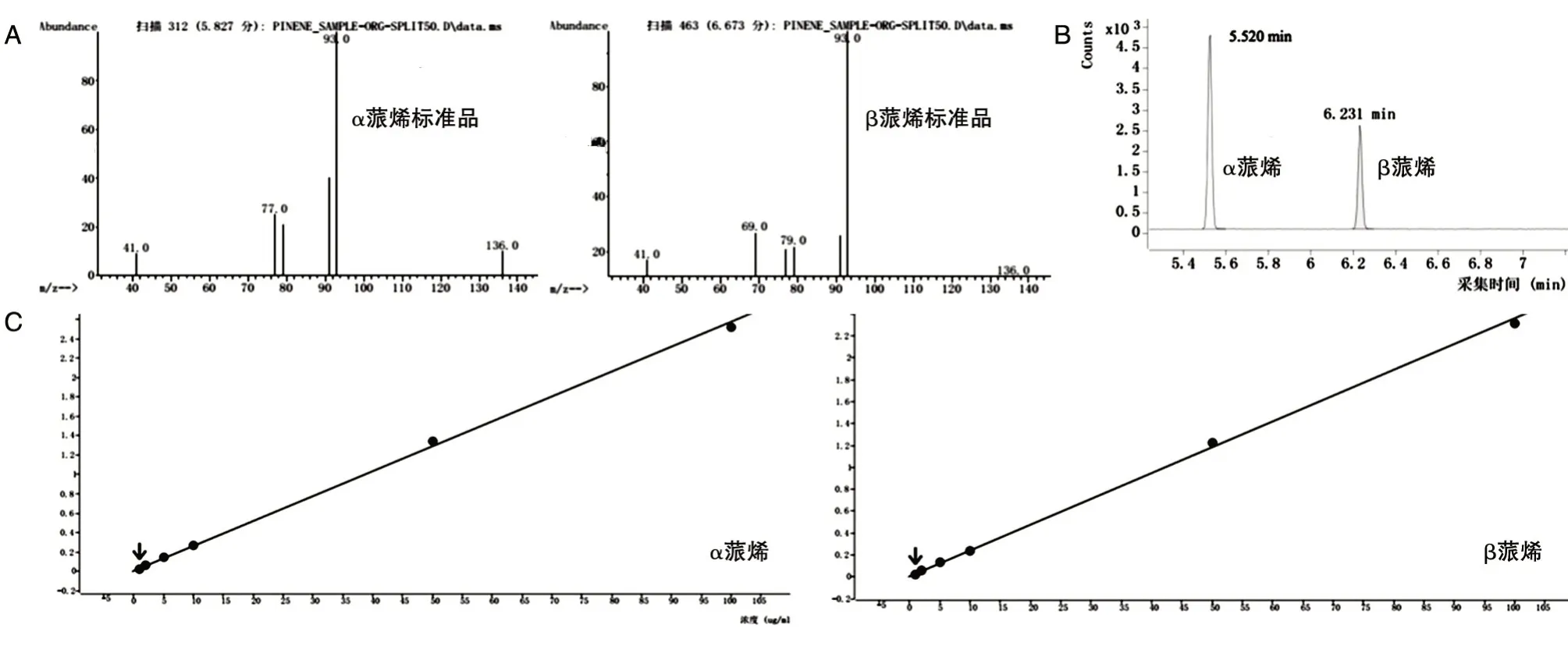
图2 GC-MS 检测蒎烯含量
2.3 目的蛋白融合表达可显著提高蒎烯产量
将工程菌E.hzh.01 和E.hzh.02 分别接种至含相应抗生素的100 mL 高密度培养基中,待菌液D600nm值为0.8~1.0,加入IPTG 并覆盖20%的正十二烷溶液,30℃、200 r/min 诱导培养72 h,分别取培养液上层十二烷层和下层菌液(水)层,用GC-MS检测蒎烯含量,结果表明十二烷层中蒎烯含量是菌液层的几十倍,因此后续研究只取上层十二烷层用于蒎烯含量检测。进一步检测得到E.hzh01和E.hzh02 的蒎烯产量分别为0.85 和1.86 mg/L,表明蛋白融合表达可显著提高蒎烯产量。
2.4 MVA上游表达载体pACYCDuet-mvaE/S的鉴定
设计引物克隆MVA 上游途径基因mvaE、mvaS,通过酶切连接方式将目的基因连到载体pACYCDuet-a 中,酶切和测序结果表明成功构建了MVA 上游表达载体pACYCDuet-mvaE/S(图3A)。将表达载体pACYCDuet-mvaE/S 转化大肠杆 菌BL21(DE3),诱导培养后SDS-PAGE 验证MVA 上游基因的表达(图3B)。mvaE和mvaS表达的蛋白相对分子质量分别为86.7×103、42.2×103,与预期相符。进一步设计引物克隆GPPS-PS融合表达基因,将目的基因酶切连接到pACYCDuet-mvaE/S 载体中,酶切鉴定结果显示成功构建了表达载体pACYCDuet-mvaE/S-GPPS-PS(图3C),转化大肠杆菌BL21(DE3)后诱导培养,SDSPAGE 验证载体中目的基因的表达(图3D),结果与预期相符。
2.5 MVA下游表达载体pET24a-ERG12-8-19-IDI的鉴定
分别利用多顺反子模型原理和BioBrick 方法将ERG12、ERG8、ERG19、IDI基因连入载体,酶切鉴定并测序显示成功构建了MVA 下游4 基因共表达载体pET24a-ERG12-8-19-IDI(图4A)和pET30a-ERG12-8-19-IDI(图4C)。将pET24a-ERG12-8-19-IDI(图4B)和pET30a-ERG12-8-19-IDI(图4D)分别转化大肠杆菌BL21(DE3)后诱导培养12 h,取全菌进行SDS-PAGE,结果表明目的基因蛋白能够正常表达。

图3 表达载体酶切鉴定和诱导表达(2)

图4 表达载体酶切鉴定和诱导表达(3)
2.6 工程菌产蒎烯能力检测
将pACYCDuet-mvaE/S-GPPS-PS 与pET24a-ERG12-8-19-IDI 或pET30a-ERG12-8-19-IDI 载体共转化大肠杆菌,构建得到的工程菌E.hzh03 和E.hzh05 分别接种至含相应抗生素的高密度培养基中,待菌液D600nm值为0.8~1.0,加入IPTG 诱导,再在培养基上覆盖20%的十二烷溶液,继续培养72 h。取十二烷层并利用GC-MS 方法检测菌株产蒎烯能力。结果表明,E.hzh03 和E.hzh05 产蒎烯能力分别为1.86 和19.26 mg/L。
3 讨论
蒎烯是重要的天然单帖类平台化合物,被广泛用于合成高能燃料、药品、香料和芳香醇等产品,在军事、药业、农业和工业等领域均有重要的应用价值。目前,在工业上制造蒎烯的途径是采用高效精馏塔从脂松节油或粗硫酸盐松节油中分离提取,存在设备及技术要求高、能耗大,分离产物纯度不高等缺点。另一方面,工业上获取松节油会消耗大量松属林木资源,造成了大量不可再生自然资源的浪费。随着系统生物学与合成生物学技术的快速发展,以大肠杆菌等底盘生物构建“微生物工厂”合成萜类化合物,已经被证明是一种可持续获取蒎烯的新途径。本研究以大肠杆菌为底盘细胞,将来源于酿酒酵母和粪肠球菌的外源杂合MVA 代谢途径基因与来源于北美巨冷杉的GPPS和PS基因共同整合到大肠杆菌中,构建了高效合成蒎烯的“微生物工厂”。
IPP 和DMAPP 是代谢合成蒎烯的必需前体物质,大肠杆菌可通过其自身的DXP 代谢途径合成IPP 和DMAPP,再将优化的GPPS和PS基因转化到大肠杆菌中实现合成蒎烯。本研究分别构建了GPPS和PS的共表达和融合表达工程菌株,利用GC-MS 检测蒎烯产量进行比较,发现融合表达的GPPS 和PS 蛋白可显著提高蒎烯在大肠杆菌中的合成效率。利用生物信息学软件分析融合蛋白的三级结构,发现GPPS 和PS 蛋白融合表达时,2 个酶的催化中心相互紧邻且成面对面的结构。与蛋白共表达比较,显示蛋白融合表达在降低蛋白毒性的同时提高了蒎烯合成酶的催化速率,降低了底物对酶的反馈抑制作用。
已有研究表明,将外源杂合的MVA 代谢途径导入大肠杆菌可提高蒎烯前体物质IPP 和DMAPP的积累,从而提高大肠杆菌合成蒎烯的效率。本研究选择的外源杂合MVA 代谢途径共有6 个酶参与代谢反应,其中上游途径为来源于粪肠球菌的mvaE和mvaS基因,这2 个基因存在于同一个基因簇上;下游途径为来源于酿酒酵母菌的ERG12、ERG8、ERG19和IDI基因,这4 个基因分布在酵母基因组不同的位置上。我们运用2 种策略构建MVA 下游代谢途径表达载体,策略一是利用多顺反子模型的原理,将基因ERG12、ERG8、ERG19和IDI体外拼接成串联序列,再由1 个启动子控制4 个基因的表达,其中每个基因含有各自的SD 序列和翻译起始终止信号;策略二是根据合成生物学中的BioBrick 方法,用同尾酶(SpeⅠ和XbaⅠ)将基因序列快速组装拼接,其中每个基因都是一个独立的操纵子模型,其蛋白独立表达。进一步的实验结果表明,将外源MVA 代谢通路和GPPS-PS融合基因共同转化大肠杆菌能够显著提高蒎烯的产量,而多顺反子模型原理构建的MVA 代谢通路具有更高的蒎烯合成效率。虽然BioBrick 方法更有利于基因组装,具有一定的优势,但后续组装的基因的表达强度随着体系的增加而减弱,一定程度上不利于含多酶活性系统的组装,从而限制蒎烯的代谢合成,而利用多顺反子模型构建的外源杂合MVA 代谢途径则更有利于大肠杆菌中蒎烯的合成。
本研究比较了GPPS、PS共表达和融合表达条件下蒎烯的合成效率,证明GPPS和PS融合表达更有利于蒎烯合成。进一步比较了不同策略下构建的外源杂合MVA 代谢途径结合GPPS-PS融合蛋白产蒎烯的能力,构建得到了蒎烯产量大幅提高的工程菌株。后续通过对发酵培养工艺的进一步优化改良,有望为今后蒎烯的大规模工业化微生物合成发展奠定一定的基础。
猜你喜欢
华人时刊(2022年9期)2022-09-06
安徽农业科学(2022年1期)2022-02-14
昆明医科大学学报(2020年12期)2021-01-26
中国生殖健康(2020年4期)2021-01-18
华人时刊(2020年15期)2020-12-14
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
中国学术期刊文摘(2016年1期)2016-02-13
中国学术期刊文摘(2016年1期)2016-02-13
