
1例马凡综合征患者原纤维蛋白1基因突变分析
2019-09-23 03:44:58倪二茹肖晶晶吴少鸿李欣谢华斌姜萍萍
山东医药 2019年23期
倪二茹,肖晶晶,吴少鸿,李欣,谢华斌,姜萍萍
(1厦门大学附属心血管病医院,福建厦门361004;2厦门大学医学院;3深圳美因医学检验实验室)
马凡综合征(MFS)是一种常染色体显性遗传性结缔组织疾病[1],它的发生与多种基因的突变密切相关[2],主要是人原纤维蛋白1基因(FBN1)[3]和转化生长因子β受体(TGFBR)基因[4]。其中,以FBN1基因突变与马凡综合征相关性的研究最多[5]。FBN1基因突变的类型多样,新的突变位点仍不断被发现,目前已报道的FBN1突变超过1 800种[6]。FBN1基因突变的类型和位点与临床表型有直接的关系,通过基因测序的方法分析FBN1基因的突变情况,有助于马凡综合征的分子诊断和家系遗传分析,同时可以研究FBN1基因突变与临床表型之间的关系[7, 8]。2018年11月,我们对1例临床确诊马凡综合征患者的FBN1基因进行检测和分析,在分子水平上探讨该患者致病的可能原因。
1 资料与方法
1.1 临床资料 选择同期厦门大学附属心血管病医院收治的1例患有马凡综合征的先证者及其所有家系成员。先证者男性,18岁,于12岁时体检发现心脏杂音,无明显不适,身高体长且明显高于同龄人,胸部外凸畸形,CT检查发现主动脉根部增宽,心脏超声提示二尖瓣、三尖瓣脱垂并关闭不全(见图1),临床诊断为马凡综合征,未予特殊处理。后患者定期复查,瓣膜反流逐渐加重。14岁时先证者心脏超声检查示左心室内径(LVD)63 mm,左心室射血分数(EF)70%,二尖瓣叶脱垂合并重度关闭不全,三尖瓣叶脱垂合并轻度关闭不全。后先证者于14岁时接受全麻下二尖瓣替换手术,术中探查见二尖瓣瓣叶增厚,键索纤细延长,二尖瓣瓣叶脱垂,瓣环扩张,瓣口对合不严。术中进行了双叶机械瓣间断缝合,完成二尖瓣置换术。测试瓣膜启闭良好。术后患者恢复良好,复查未见异常。本研究符合赫尔辛基宣言,并得到了厦门大学附属心血管病医院伦理委员会的批准。先证者及其所有家系成员均签署了知情同意书。先证者及其所有家系成员的个人信息均得到严格的保密。
1.2 家系情况 该先证者家系成员2代共3人,其中患病者1例(即先证者,男性),见图2。先证者的父母无马凡综合征的临床症状,先证者无兄弟姐妹,父母双方无相关症状的亲属。
1.3 基因组DNA提取 经先证者及其所有家系成员同意,取先证者及其父母外周静脉血4 mL,利用全血基因组提取试剂盒(HiPure Tissue & Blood DNA Kit,美基生物)提取基因组DNA,并测定浓度。
1.4 目标区域捕获测序 采用目标区域捕获测序技术对先证者进行基因检测。具体步骤如下:采用Agilent定制化探针试剂盒(瑞士Roche公司)建立含目标基因的全基因文库,包括已知的13种遗传性心源性猝死相关疾病的88个基因,其中包括马凡综合征相关基因(FBN1、TGFBR1和TGFBR2)。采用Illumina HiSeq2500测序仪(美国Illumina公司)进行高通量测序。
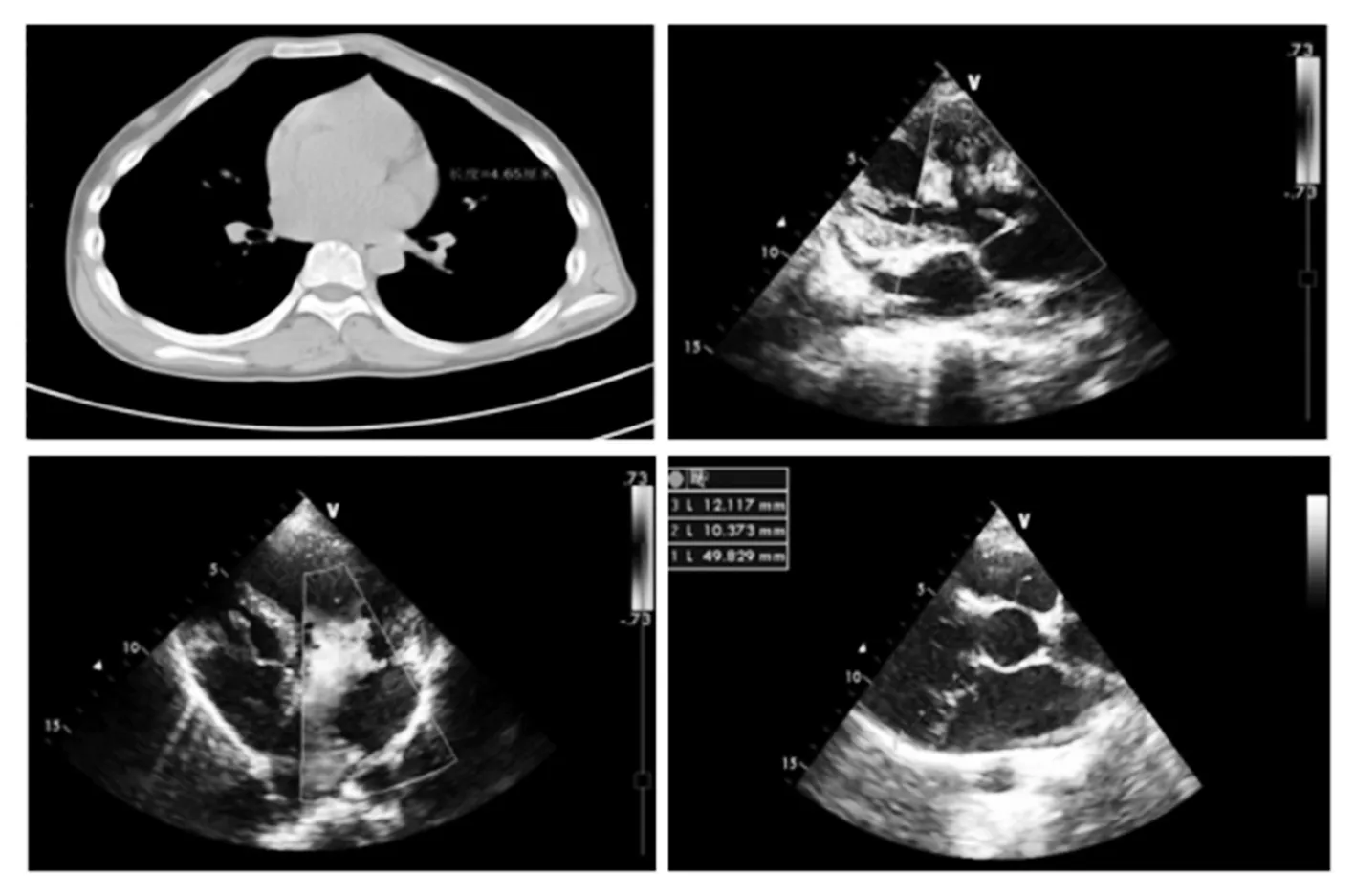
注:左上为CT示主动脉根部宽度为4.46 cm;右上为心脏彩超示三尖瓣反流;左下为心脏彩超示二尖瓣反流;右下为心脏彩超示二尖瓣脱垂。
图1 先证者心脏超声表现
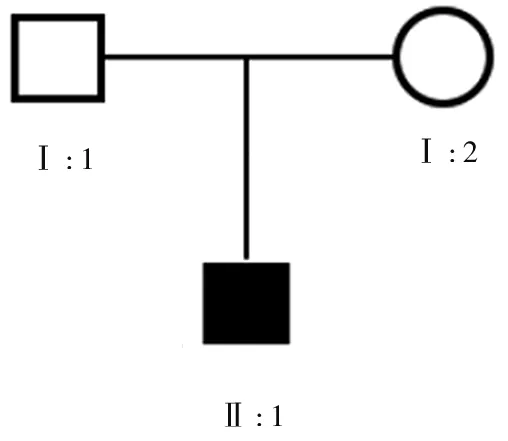
注:Ⅰ:1、Ⅰ:2分别为先证者的父母;Ⅱ:1为先证者。
图2 先证者的家系图
1.5 生物信息学分析 对上述测序获得的原始数据采用BWA软件(http://biobwa.sourceforge.net)与hg19人基因组数据库(http://hgdownload.cse.ucsc.edu/)进行比对,之后使用GATK软件(https://software.broadinstitute.org/gatk/)进行变异分析,变异位点经深圳美因临床检验实验室自研的软件进行注释。变异注释工作包括:识别单核苷酸变异(SNV)、插入和缺失变异(INDEL)是否引起蛋白质编码或者是氨基酸改变,识别变异是否在保守区域;识别变异在人群数据库中的频率,数据库包括国际千人基因组计划t(http://browser.1000genomes.org/)、美国国家心肺和血液研究所外显子组测序计划(https://evs.gs.washington.edu/EVS/)、外显子组整合联合数据库(ExAC,http://exac.broadinstitute.org/)和Dbsnp数据库(https://www.ncbi.nlm.nih.gov/snp/)等;计算突变有害性分数;查找变异与疾病之间的关系,用到的数据库包括HGMD(http://www.hgmd.cf.ac.uk/ac/index.php)、OMIM(https://omim.org/)、NCBI(https://www.ncbi.nlm.nih.gov/)和CGD(http://www.candidagenome.org/)。对数据库中的纯合变异以及最小等位基因频率(MAF)大于0.01进行过滤,进一步筛选先证者样本中的DNA变异。
1.6 位点解读 数据解读规则参考美国医学遗传学和基因组学学院(ACMG)相关指南。
1.7 Sanger测序 经目标区域捕获测序筛选的候选变异位点采用Sanger测序进行验证,同时在先证者父母中进一步验证。具体操作步骤如下:聚合酶链反应(PCR),反应条件为94 ℃预变性3 min;94 ℃ 30 s、56 ℃ 45 s、72 ℃ 60 s,30个循环,72 ℃延伸5 min;将PCR产物送至赛默飞世尔科技(中国)有限公司进行Sanger测序。
1.8 蛋白结构分析 将突变后的DNA序列进行氨基酸序列预测,之后使用SWISS-MODEL找到与目标序列一致度≥30%的已知结构作为模板。
2 结果
2.1 目标区域捕获测序 经目标区域捕获测序,共产出773.80 Mb数据,比对率达99.83%,目标区域平均测序深度为523.53x,覆盖度为99.90%。其中FBN1 TGFBR1和TGFBR2基因的测序深度分别达542x、516x、593x,覆盖度均为100%。共检测出单核苷酸变异位点(包括错义突变和同义突变)124个;插入和缺失变异2个。通过对公共数据库过滤,频率小于0.01的罕见突变有3个。
2.2 生物信息学分析 生物信息学技术分析发现先证者携带FBN1基因c.2023_2026delTTTG突变。转录本NM_000138.4,氨基酸变异p.F675Vfs*42,杂合突变,未在千人基因组数据库及EXAC数据库中发现突变频率。该突变位于17号外显子区,属于移码突变,导致了675位的苯丙氨酸变成了缬氨酸,并产生新的阅读框架,终止于第675位密码子下游42位密码子处,因此会生成一个缩短并变异的RNA以及蛋白。
2.3 Sanger测序 采用Sanger测序对FBN1基因移码突变的先证者及其父母进行验证,结果显示,先证者为FBN1(c.2023_2026delTTTG,p.F675Vfs*42)的杂合移码突变,与目标区域捕获测序发现的FBN1基因突变一致,而其父母不具有该突变,因此该突变为新生突变(denovo突变)。见图3。
2.4 蛋白结构分析 建立FBN1基因p.F675Vfs*42突变翻译的异常蛋白3D构象后,显示移码突变导致了675位的苯丙氨酸变成了缬氨酸,并产生新的阅读框架,终止于第675位密码子下游42位密码子处。FBN1编码的正常的FBN1前体由2871个氨基酸组成,而p.F675Vfs*42突变编码的只有716个氨基酸,缺失了大部分的原FBN1正常结构,彻底改变了蛋白质的构象,损坏了蛋白质的功能,不能转化成正常的FBN1。
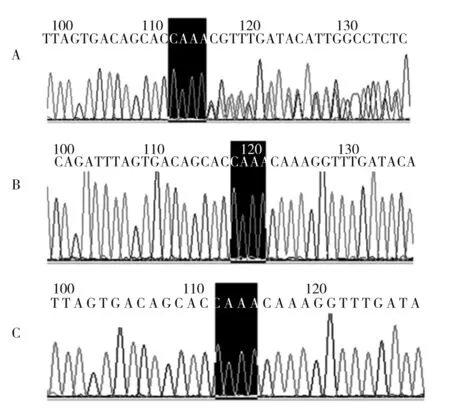
注:A为患者Sanger测序结果,存在c.2023_2026delTTTG突变,杂合缺失;B为患者父亲Sanger测序结果,无突变;C为患者母亲Sanger测序结果,无突变。
图3 先证者及其父母FBN1基因的Sanger测序峰图
3 讨论
马凡综合征是一种常染色体显性遗传的疾病,常表现为蜘蛛指(趾)病、细长肢体病、长肢病、指(趾)过长综合征、长瘦、抑郁症、蜘蛛手合并晶状体脱位等[1]。在遗传学上,马凡综合征具有外显率高、表现度不一等特点,故临床表现多种多样[9, 10],主要变现为骨损坏、眼损害及心血管病变三联征。该病属于结缔组织遗传缺陷疾病,其病变可累及全身各个系统,而严重的心血管并发症是患者的主要致死因素[11]。根据修订版Ghent诊断方案[12],若患者无马凡综合征家族史,但主动脉根部直径Z值≥2,合并晶状体脱位、FBN1基因突变,系统评分>7分,三者之一;或晶状体脱位合并FBN1基因突变并主动脉病变,马凡综合征诊断成立。本文先证者临床症状表现胸廓畸形、臂展和身高均过大,心脏彩超检查示主动脉根部增宽,二尖瓣、三尖瓣脱垂并关闭不全,具有典型的马凡综合征的临床表现。
FBN基因家族和TGFBR基因家族是马凡综合征的主要致病原因。FBN基因家族中FBN1基因编码的人FBN1位于15号染色体的长臂(15q15~q21.1),包含65个外显子,将近10 000个核苷酸,编码的蛋白质相对分子量约为350 kDa。该蛋白含有47个表皮生长因子样结构域,7个8-半胱氨酸结构域,2个杂交结构域和1个富脯氨基酸结构域。FBN1蛋白与其他细胞外基质形成弹性纤维,维持组织的弹性。本文在对先证者的基因组DNA进行目标区域捕获测序后,分析了马凡综合征相关的FBN1、TGFBR1和TGFBR2基因的突变情况。经过生物信息学技术的分析,发现先证者携带了FBN1基因c.2023_2026delTTTG突变。该突变位于17号外显子区,由于移码突变,导致了675位的苯丙氨酸变成了缬氨酸,并产生新的阅读框架,终止于第675位密码子下游42位密码子处,因此会生成一个缩短并变异的RNA以及蛋白。进一步的蛋白结构分析发现正常FBN1基因编码的FBN1蛋白有2 871个氨基酸,而FBN1基因c.2023_2026delTTTG突变编码的FBN1蛋白只有716个氨基酸,缺失了大部分的FBN1蛋白的正常结构,彻底改变了蛋白质的构象,损坏了蛋白质的功能,不能转化成正常的FBN1蛋白。根据ACMG评估指南,定义该位点为疑似致病突变的变异,而GeneDx基因检测公司认为该位点为致病突变。Howarth等[13]在2007年首次报道了该突变,其在两个非亲属关系的马凡综合征的患者中检测到了该突变;并且在家系验证中,10个未患病人员未检测到该突变,而在5例患病人员中均检测到了该突变;且未在各大数据库中报道。除此之外,FBN1基因c.2023_2026delTTTG突变尚未在其他文献中报道过。
本文的先证者父母没有携带相同的突变,而先证者没有兄弟姐妹,故无法对该基因型与临床表型的关系进行验证。虽然没有该位点与马凡综合征的表型关联的文献研究报道,但是在与马凡综合征相关的FBN1基因突变中,提早终止密码突变(PTC)以及移码突变是FBN1突变中的最主要的两种类型[14],PTC突变与严重的骨骼和皮肤表型相关[15],很少出现眼部的病变。
综上所述,本文通过对1例临床确诊马凡综合征的先证者及其父母进行FBN1基因的检测,发现先证者携带的FBN1基因c.2023_2026delTTTG突变会产生一个截短的FBN1蛋白,可能是马凡综合征的一个致病突变。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
临床输血与检验(2022年3期)2022-06-22 02:52:50
中国生殖健康(2020年2期)2021-01-18 02:51:26
趣味(数学)(2020年4期)2020-07-27 01:44:16
支部建设(2020年15期)2020-07-08 12:34:32
郑州大学学报(医学版)(2019年3期)2019-06-03 06:19:32
传染病信息(2019年2期)2019-05-17 13:16:04
小学生导刊(2018年13期)2018-06-29 03:49:00
百科知识(2015年18期)2015-09-10 07:22:44
重庆医学(2015年12期)2015-03-05 05:52:54
