
基因BSCL2新发突变致全身脂肪营养不良1例
2019-08-22 08:49:26付东霞刘芳卫海燕
中国现代医学杂志 2019年15期
付东霞,刘芳,卫海燕
[郑州大学附属儿童医院(河南省儿童医院),河南 郑州 450000]
1 临床资料
患者,女性,年龄1岁1个月,13 d前因发热2 d入住河南省儿童医院新生儿内科。住院期间发现血糖、血脂升高,甘油三酯22.5 mmol/L,较正常儿童升高,同时伴高胰岛素血症,结合患儿疾病特点,查阅相关资料,考虑为先天性全身脂肪营养不良。
入院后给予护肝、胰岛素控制血糖、低脂饮食及补液等对症支持治疗。10 d后精神反应好转,血糖平稳,甘油三酯下降出院。
患儿出院后给予低脂饮食,并随诊。监测血糖正常,转氨酶及血脂好转;腹部彩超提示肝脏及肾脏进行性增大。1个月时逐渐出现腹胀、消瘦,2个月时出现皮下脂肪逐渐消失,肌肉肥大;3~4个月出现全身皮肤色素沉着,以颈部及腋下明显;5~6个月出现全身皮毛增多增粗,以毛发浓密显著,同时伴毛发卷曲;随年龄增长,腹部膨隆渐明显,肝脏进行性增大。见图1。予基因检测。
结果显示,该患儿基因突变位置为chr11:62460139,为一处纯合突变:c.567_568delGA,导致氨基酸改变p.Glu189fs(移码突变),父母为杂合突变。该突变位点未见报道,为新发突变。参考美国医学遗传学和基因组学协会相关基因突变解读指南,该位点为恶性突变位点,有很强可能性致病。
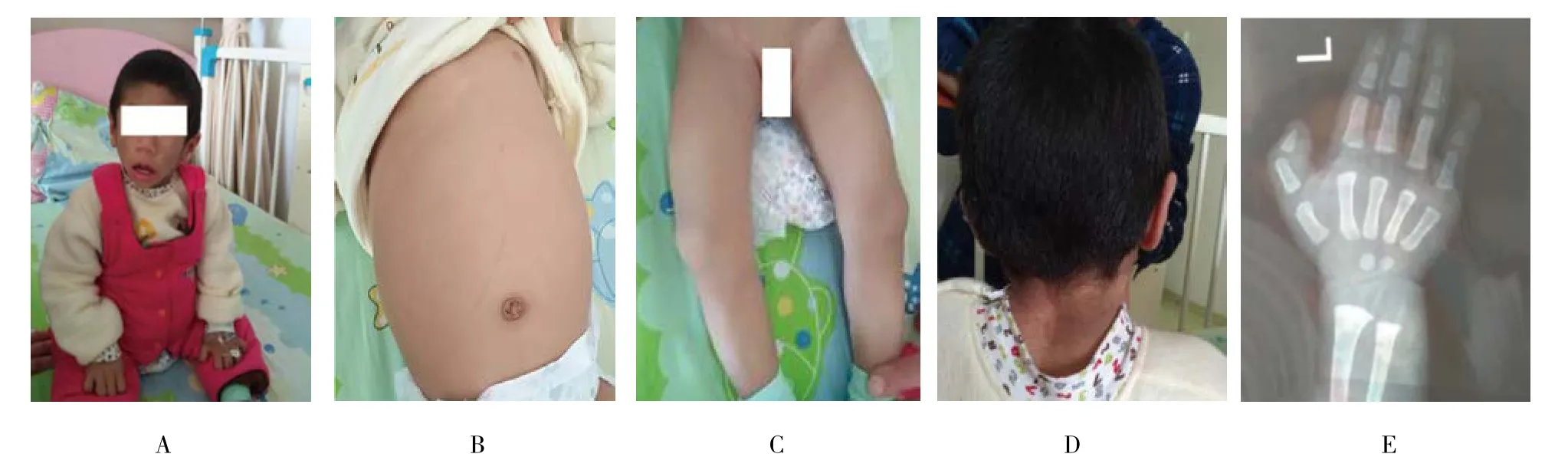
图1 患者随访资料
2 讨论
先天性全身脂肪营养不良(congential generalized lipodystrophy,CGL)是由 BERARDINELLI[1]于 1954 年首先报道,1959年SEPI[2]报道,故本病又名Berardinelli-Seip综合征。是非常少见的一种遗传代谢疾病,全球发病率约为1/1 000万[3]。主要特点为出生时脂肪组织几乎完全消失[4]、极度胰岛素抵抗、高雄激素血症、高甘油三酯血症、脂肪肝、甚至肝硬化、黑棘皮病[5]。
本研究中,患者无CGL家族史,但生后即有全身脂肪消失、高脂血症、胰岛素抵抗,进一步完善基因检测提示为CGL,随着病情进展出现食欲亢进、多毛、色素沉着、智力低下等表现,符合CGL表现。CGL的病因复杂。已发现的致病基因至少有4种,95%的CGL与AGPAT2和BSCL2基因突变相关[6]。BSCL2基因位于染色体11q13上,编码398个氨基酸的跨膜蛋白。在脑组织中广泛表达,因此智力损害较为常见。该患儿存在BSCL2基因突变,且突变位点未见报道,为新发突变。故该患儿存在全身脂肪营养不良、智力损害可以解释。
CGL的主要治疗方法是降低高甘油三酯血症和心血管风险。饮食管理是非常重要的,鼓励CGL患者遵循高碳水化合物,低脂肪的饮食。
猜你喜欢
英语世界(2023年6期)2023-06-30 06:29:10
老年人(2022年8期)2022-04-29 00:44:03
锦州医科大学报(2021年8期)2021-11-18 09:11:53
中老年保健(2021年9期)2021-08-24 03:51:56
中国生殖健康(2020年2期)2021-01-18 02:51:26
基层中医药(2020年9期)2020-11-27 01:55:26
祝您健康(2018年12期)2018-11-27 02:30:34
小学生导刊(2018年13期)2018-06-29 03:49:00
小猕猴智力画刊(2017年10期)2017-11-06 12:57:51
食品工业科技(2014年13期)2014-03-11 18:17:10
