
气相色谱-质谱法测定食用植物油中3-氯-1,2-丙二醇酯的不确定度评定
2019-07-31 08:06熊丽周鸿
实验与检验医学 2019年4期
熊丽,周鸿
(江西省疾病预防控制中心,江西 南昌 330029)
氯丙醇酯是近年来国际上较为关注的新型食品污染物,以精炼植物油中3-氯-1,2-丙二醇酯(3-MCPD酯)污染最为显著,其含量是游离态氯丙醇(3-MCPD)的上百甚至上千倍[1]。近年来毒理研究表明3-MCPD酯具有一定的毒性作用[2],但对其健康的担心主要是因为氯丙醇酯会在加热、加酸、微生物或人体肠道胰脂酶的作用下水解成游离态的氯丙醇[3],而游离氯丙醇(3-MCPD)是公认的食品污染物,具有潜在的致癌性、神经毒性、免疫毒性、遗传毒性和生殖毒性[4],会对人体健康构成极大威胁。
不确定度是与测量结果相关联的参数,表征合理地赋予被测量的分散性。为了保障实验室提供的分析数据准确、可靠,及时分析与评定结果的不确定度具有十分重要的意义。本研究依据JJF 1059.1-2012《测量不确定度评定与表示》[5],参照《2016年国家食品污染物和有害因素风险监测工作手册》中《食品中脂肪酸氯丙醇酯含量的标准操作程序》及相关文献[6-8],采用气相色谱-质谱法,对食用植物油中3-氯-1,2-丙二醇酯测量结果的不确定度进行了评定,为进一步探讨测量结果的准确程度和方法的可靠性提供参考。
1 材料与方法
1.1 仪器与试剂 7890B-7000C气相色谱-质谱联用仪:美国Agilent公司;XP-205电子天平:瑞士梅特勒-托利多公司;SK8200HP超声清洗器:上海科导超声仪器有限公司;OSB-2100旋转蒸发仪:日本ELELA公司;DHG-9101-3SA电热恒温箱:上海鸿都电子科技有限公司;气密针(1.0ml):瑞士hamilton公司。
3-氯-1,2-丙二醇棕榈酸二酯(3-MCPD 酯)、d5-3-氯-1,2-丙二醇棕榈酸二酯 (d5-3-MCPD酯)加拿大TRC公司。正己烷、甲醇、乙酸乙酯(色谱纯):德国Merk公司;甲基叔丁基醚(色谱纯):上海国药沃凯;七氟丁酰基咪唑:日本TCI公司;乙醚(分析纯):国药集团。硅藻土基质固相分散萃取柱、高效脱水剂:福州勤鹏生物科技有限公司。
1.2 方法
1.2.1 色谱条件 DB-5MS毛细管色谱柱 (30m×0.25mm×0.25μm):美国 Agilent公司;进样口温度:280℃; 载气: 高纯氦气 (纯度≥99.999%); 流速1.0ml/min;程序升温:50℃保持 1min,再以 2℃/min的速度升至90℃,最后以40℃/min的速度升至280℃,保持5min;进样方式:不分流进样;进样体积:1.0μl。
1.2.2 质谱条件 电离方式为电子轰击源 (EI);电离能量 70ev;溶剂延迟 3min;阱温度:230℃;传输线温度:250℃;监测方式选择离子扫描(SIM);3-MCPD 衍生物定性离子 m/z为 253、289、453,其中253为定量离子;内标物d5-3-MCPD衍生物定性离子m/z为257、294、278,其中257为定量离子。
1.2.3 样品前处理 准确称取 0.1g(精确至 1mg)食用植物油于50ml离心管中,加入100μl(浓度为2.0μg/ml) 的内标使用液, 加入 0.5ml甲基叔丁基醚/乙酸乙酯溶液,混匀,超声;水解:加入 1.0ml甲醇钠溶液水解后加入冰乙酸中和以终止水解;净化:用硅藻土小柱(基质分散固相萃取法)净化,先以正己烷淋洗,再以无水乙醚洗脱,洗脱液充分脱水后转移至鸡心瓶中,减压浓缩至近干,用滴管将其转入带磨口塞的5ml玻璃刻度试管中,再用正己烷分两次充分洗涤鸡心瓶内壁,合并洗涤液至约为 1.0ml;衍生:用气密针加入 50μl七氟丁酰基咪唑 (HFBI)后于 75℃的电热恒温箱中衍生30min;衍生结束后,取出冷却至室温,补加正己烷至体积为1.0ml,再入2ml饱和氯化钠溶液,充分涡旋,放置后取上层有机相以无水硫酸钠脱水后供气相色谱-质谱进样分析。
1.2.4 标准曲线的制备 以0.10g饱和氯化钠为本底,分别准确移取 10、50、100、200、300μl 3-MCPD酯标准使用液 (浓度为2.0μg/ml) 于50ml离心管中, 分别相当于 20、100、200、400、600ng的氯丙醇酯,以样品为0.1g计算,相当于氯丙醇酯浓度分别为 0.200、1.00、2.00、4.00、6.00mg/kg (均以氯丙醇计),再各加入内标工作液(2.0μg/ml)100μl,混匀,之后步骤同1.2.3样品前处理。
1.2.5 样品检测与计算 以3-MCPD衍生物的峰面积与内标衍生物的峰面积之比(Y)对3-MCPD酯的质量(X,ng)进行线性回归,得到标准工作曲线,由标准工作曲线计算氯丙醇酯的质量,按下式计算食用植物油中3-MPCD酯的含量。
式中:X——试样中氯丙醇酯含量 (均以游离态氯丙醇计),单位为mg/kg;
A——由标准曲线计算试样中氯丙醇酯的质量,单位为ng;
m——食用油的取样量,单位为g;
2 不确定度的来源
通过对实验过程进行分析,不确定度的来源主要有重复性(随机效应)、回收率(样品前处理)、样品称量、内标加入量、标准溶液的配制以及标准曲线的拟合等引入的。不确定度因果图见图1。
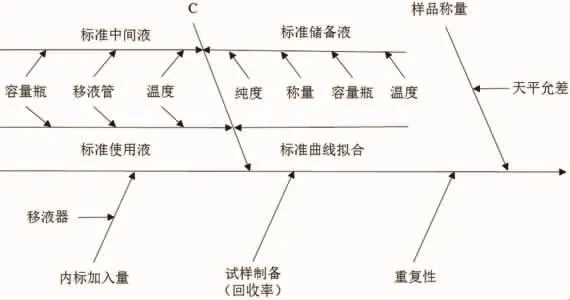
图1 不确定度因果图
3 不确定度评定
3.1 测定的重复性(Rep)引入不确定度 对同一食用植物油样品进行了6次平行测定,测定结果分别 为 0.775、0.688、0.781、0.702、0.788、0.825mg/kg,平均浓度0.760mg/kg,根据贝塞尔公式进行不确定度计算:
重复性的相对不确定度:Urel(rep)=×100%=×100%=2.87%
3.2 回收率(R)引入的不确定度
根据贝塞尔公式,
回收率相对不确定度:Urel(R)=×100%,Urel(R)=2.69%
用检测统计数据t对平均回收率进行显著性检验,在95%置信概率下,当t值大于或等于t的临界值时,则回收率与1之间有显著性差异,要引入修正因子,当t值小于t的临界值时,回收率与1之间无显著性差异,可以不做回收率校正。

在95%置信概率,自由度n=5时的t分布临界值 t0.95(5)=2.57,本研究 t<t0.95(5),回收率和 1 之间无显著性差异,可以不做回收率校正。见表1。

表1 回收率测定结果
3.3 样品称量(W)引入的不确定度 称取0.1g样品,据天平检定证书其最大允许误差为±0.05mg,按均匀分布处理(k=),则称重不确定度为:u(w),u(w)0=0.029mg
该标准不确定度需计算两次,一次为皮重,一次为总重,则称量带入的标准不确定度为:u(w)=,u(w)=0.041mg
称量引入的相对不确定度:Urel(w)=×100%=0.041%
3.4 加入内标使用液带入的不确定度(V) 用量程为 100μl移液器吸取 100μl内标使用液(2.0μg/ml),根据 JJG 646-2006《移液器检定规程》[9]规定100μl可调移液器吸取100μl时容量最大允许误差为±2.0%,按矩形分布,(k=),
内标引入相对标准不确定对为:Urel(v)=×100%=1.15%
3.5 样品溶液中3-MCPD酯测定浓度(c)带入不确定度 样品溶液中3-MCPD酯测定浓度(c)带入不确定度由标准品的纯度、标准溶液的配制以及标准曲线拟合三部分带入。
3.5.1 标准品引入的不确定度 3-氯-1,2-丙二醇棕榈酸二酯标准品,纯度为 98.0±0.5%,均匀分布,
标准品带入相对标准不确定度为:Urel(c)0=×100%=0.30%
3.5.2 标准溶液配制带入的不确定度 标准溶液配制带入的不确定度由3-MCPD酯标准储备液、中间液及使用液的配制三部分带入。
3.5.2.1 标准储备液配制带入的不确定度 准确称取3-MCPD酯标准品10mg于10.0ml容量瓶中,用乙酸乙酯溶解并定容,浓度1.0mg/ml。标准储备液配制带入不确定度包括三部分,天平称量(m)、容量瓶(v)、及温度变化(T)。
天平称量(m):根据天平的检定证书,称量的最大允许误差为±0.05mg,称重有两次称量获得(一次为皮重,一次为毛重),因每次都是独立称量结果,故计算两次。u(m)0===0.029mg,u(m)==0.041mg
天平称量引入的相对不确定度:Urel(m)=×100%=0.41%
容量瓶(v):10ml容量瓶(根据 JJG 196-2006[10],A 级允差±0.02ml),按矩形分布:u(v)=×100%=0.12%
温度变化 (T):由于温度波动引起的体积变化,温度波动范围为 (20±5)℃,呈均匀分布,k=,乙酸乙酯的体积膨胀系数为 1.38×10-3℃-1,产生的体积变化 V=±(10×1.38×10-3×5)=±0.069ml,u(T)=×100%=0.40%
配制标准储备液带入的不确定度:Urel(c)1==0.59%
3.5.2.2 标准中间液配制带入的不确定度 用单标线移液管吸取1.0ml标准储备液于50.0ml容量瓶中,正己烷定容,浓度为20.0μg/ml。标准中间液配制带入不确定度包括三部分,移液管(v0)、容量瓶(v)、及温度变化(T)。
50ml容量瓶 (根据JJG 196-2006,A级允差±0.05ml),u(v)=×100%=0.058%
温度变化 (T):正己烷的体积膨胀系数为1.36×10-3℃-1。
产生的体积变化 V=±(50×1.36×10-3×5)=±0.34ml,u(T)=×100%=0.39%。
1.0ml单标线移液管(根据 JJG 196-2006,A 级允差±0.007ml),u(v)0=×100%=0.40%。
配制标准中间液带入的不确定度为:Urel(c)2==0.59%。
3.5.2.3 标准使用液配制带入的不确定度 用单标线移液管吸取1.0ml标准中间液于10.0ml容量瓶中,正己烷定容,浓度为 2.0μg/ml。3-MCPD 酯标准使用液配制带入不确定度包括:移液管(v0)、容量瓶(v)、及温度变化(T)。
10ml容量瓶 (根据JJG 196-2006,A级允差±0.02ml),u(v)=×100%=0.12%;
温度变化 (T):正己烷的体积膨胀系数为1.36×10-3℃-1,
产生的体积变化 V=±(10×1.36×10-3×5)=±0.068ml,u(T)=×100%=0.39%;
1.0ml单标线移液管(根据 JJG 196-2006,A 级允差±0.007ml),u(v0)=×100%=0.40%
配制3-氯丙醇酯标准使用液带入的不确定度为:Urel(c)3==0.57%
3.5.2.4 标准曲线拟合带入的不确定度 对标准系列每个浓度点进行三次测定,由最小二乘法原理进行拟合求得标准曲线,Y=0.0065x-0.0017,r2=0.9997,样品测定 6 次响应值:0.505、0.478、0.518、0.490、0.521、0.559, 带 入 方 程 求 得 浓 度 (ng) 为78.0、78.4、79.9、75.6、80.5、86.3,由此求得 cc=79.0
标准曲线标准差:SA=SA=0.036,c=(20+100+200+400+600)/5=264式中,sA—标准
曲线的标准差;p—样品溶液的测定次数 (p=6);n—标准溶液的测定总次数 (n=15);b—标准曲线的斜率;a—标准曲线的截距;cc—由标准曲线求得的样品溶液中浓度;c—各标准溶液中浓度的平均值;ci—由标准曲线方程得出的标准溶液中浓度的测定值。见表2。
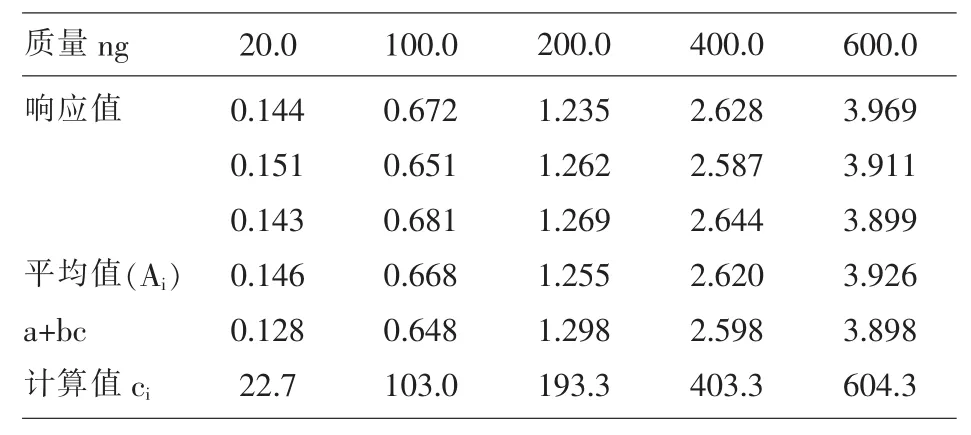
表2 标准曲线拟合带入的不确定度的参数

标准曲线拟合带来的相对不确定度:Urel(c)4=×100%=4.35%
3.5.2.5 样品溶液(c)带入总的相对标准不确定度为Urel(c)=4.47%

3.6 合成相对不确定度和扩展不确定度
3.6.1 合成不确定度
Urel(X)==6.1%
3.6.2 扩展不确定度 按国际惯例,95%置信概率下取包含因子k=2,测量结果的相对扩展不确定度为:U=Urel(X)×k×X,U=6.1%×2×0.760=0.09mg/Kg
3.7 不确定度报告:X=(0.76±0.09)mg/Kg (k=2,p=95%)
3.8 不确定度各分量结果 见表3。

表3 各分量的不确定度
4 讨论
本次评定将样品处理过程中的水解、净化、衍生等操作带来的不确定度,通过计算回收率的不确定度来表示。
根据内标法的定量原理,本次评定并没有计算内标纯度以及配制所带来的不确定度,因为只要在实验中对样品及标准系列加入同一内标标准使用液,那么内标溶液的浓度对待测组分的定量将没有影响,因此只需分析内标的加入体积所带来的不确定度。
本文建立了GC-MS法测定食用植物油中3-氯丙醇酯的不确定度评定方法,定量计算出测量结果的可信程度,评估了合成不确定度和扩展不确定度,对检测结果的符合性判定和后续的试验优化工作具有重要意义,通过比较各分量的相对不确定度大小可以看出,标准曲线拟合是不确定度最主要的来源,其次是重复性与回收率,称取样品所带来的不确定度可以忽略不计。
猜你喜欢
化工设计(2022年4期)2023-01-02
分子催化(2022年1期)2022-11-02
石油炼制与化工(2022年2期)2022-02-15
食品工业科技(2021年23期)2021-12-16
化工管理(2020年26期)2020-10-09
酿酒科技(2020年7期)2020-08-03
广西教育·C版(2020年4期)2020-07-09
中国油脂(2017年7期)2017-09-16
食品与生活(2017年5期)2017-05-27
电子技术与软件工程(2016年24期)2017-02-23
