
催化动力学光度法测定食品中的痕量硒
2019-07-25 10:22郭楠楠段秋虹
食品工业 2019年7期
郭楠楠,段秋虹
郑州科技学院,郑州市食品安全快速检测重点实验室(郑州 450064)
硒是人和动物所必需的微量元素[1],硒在自然界中存在的形式是无机硒和有机硒,近几年,含硒样品的种类越来越多,检测的手段也各有不同,中华人民共和国国家标准GB 5009.93—2017《食品安全国家标准 食品中硒的测定》中规定了食品中硒含量的测定方法,主要有氢化物原子荧光光谱法、分光光度法、荧光分光光度法和电感耦合等离子体质谱法等[2]。与这些测定方法相比,催化动力学光度法具有仪器简单、测定费用较低、灵敏度高等优点[3],因此,试验探讨了催化动力学光度法对食品中硒含量的测定条件及动力学条件,建立一种测定食品中痕量硒的新方法。
1 材料与方法
1.1 材料与试剂
黑木耳、核桃、红小豆(市售);100 mg/L硒标准溶液(用时稀释至0.1 μg/mL,中国·派尼化学试剂厂);亚甲蓝(郑州派尼化学试剂厂);溴酸钾(郑州派尼化学试剂厂);硝酸溶液(郑州派尼化学试剂厂);盐酸(郑州派尼化学试剂厂);硫酸(洛阳昊华化学试剂有限公司)。
1.2 仪器与设备
HH-S型水浴锅(0~100 ℃),郑州长城科工贸有限公司;101-OABS电热鼓风干燥箱,上海科恒实业发展有限公司,UV-4802紫外可见光分光光度计,上海悦丰仪器仪表有限公司;电子万用炉,北京市永光明医疗仪器有限公司。
1.3 试验方法
1.3.1 样品处理
将黑木耳、核桃、红小豆用水洗净,在65~70 ℃条件下,在烘箱中烘干样品,粉碎完全。用电子天平称取1.000 g粉碎好的样品于锥形瓶中,添加少量的水润湿,再加入25 mL 1:4的高氯酸-硝酸混酸,在室温下静置过夜,在锥形瓶中加入2~3粒玻璃珠,在可调式电热炉上缓慢加热,并补加硝酸,直至试样溶液呈透明。溶液冷却后,加入20 mL 1:1的硫酸溶液,后转入分液漏斗中,依次加入1.5 g的溴化钾(溴化钾需要先用少量的水溶解)、3 mL的磷酸溶液,混匀后加入25 mL的苯,振荡1 min,分层后将水相移入另一份液漏斗中,再加入25 mL的苯重复萃取一次,合并有机相,用50 mL的水反萃取硒两次,将两次的有机相合并于100 mL容量瓶中,加蒸馏水稀释至刻度。
1.3.2 样品分析方法
取两根相同的25 mL的具塞的比色管,在其中的一根中加入1.5 mL的硒标准使用液做催化反应,另外一根中不加硒标准使用液,再依次分别加入2.00 mL硝酸溶液、2.00 mL亚甲蓝溶液、2.50 mL溴酸钾溶液,再加入蒸馏水,稀释至刻度线,振摇混合均匀,置于75 ℃水浴中加热5 min,立即取出置于流水中冷却至室温。冷却至室温后,以水作参比,用1 cm比色皿在665 nm处分别测定出非催化反应溶液的吸光度A0和催化反应溶液的吸光度A,计算出ΔA。
1.4 样品中硒含量的测定
在比色管中依次加入2.00 mL硝酸、2.00 mL亚甲蓝溶液、2.5 mL溴酸钾溶液,加水稀释至25 mL测定它的吸光度,记为空白值。吸取5.00 mL经处理的样品消化液于25 mL的比色管中,再在比色管中依次加入2.00 mL硝酸溶液、2.00 mL亚甲蓝溶液、2.5 mL溴酸钾溶液,用蒸馏水稀释至刻度25 mL,轻轻地摇匀并混合均匀,将摇匀的比色管置于75 ℃的恒温水浴箱中连续加热5 min,水浴加热完成后,在冷水的作用下,将比色管置于冷水流下冷却至室温。再用紫外分光光度计测定它的吸光度。在待测样液中分别加入0.1和0.2 μg的硒标准样液,测定其吸光度,计算样品中硒的含量及回收率。
1.5 样品中硒含量的计算
根据标准曲线方程计算样品中硒浓度,根据硒的浓度计算出样品中硒的含量。

式中:X为试样中硒含量,mg/kg;ρ为试样中硒的质量浓度,mg/kg;m为试样质量,g;V1为试样用比色皿的体积,mL;V2为样品消解后定容体积,mL;V3为移取消化液的体积,mL。
2 结果与分析
2.1 波长对硒吸光度的影响
用紫外分光光度计测定不同波长情况下,溶液的不同的吸光度。在紫外分光光度计上的多波长设置中,分别设置波长655,660,665,670,675和680 nm,按1.3.2进行试验。观察试验现象并记录试验数据,具体结果见图1。催化反应与非催化反应的最大吸收波长为665 nm处,可看出在665 nm处为最适合的测定波长。
2.2 试验条件的选择及工作曲线的确定
2.2.1 酸度的选择
用吸量管分别吸取1.00,1.50,2.00,2.50和3.00 mL 0.025 mol/L的硝酸溶液于5根25 mL比色管中。按照试验方法测定其吸光度。结果见图2。硝酸的最适用量为2.00 mL,此时ΔA最大,所以试验选择硝酸的用量为2.00 mL。
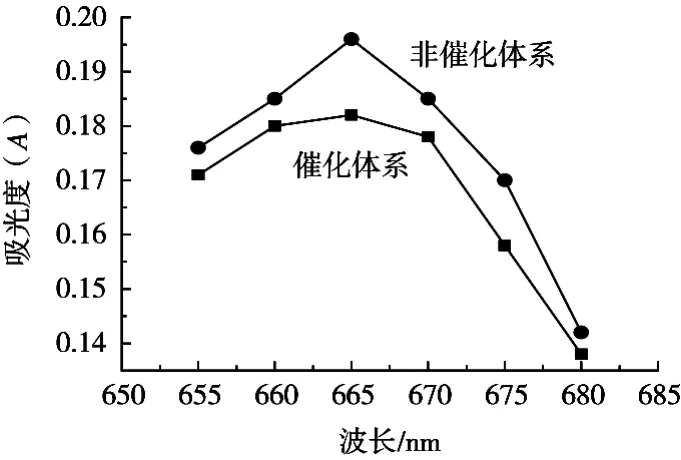
图1 吸收波长对硒吸光度的影响

图2 硝酸用量对反应的影响
2.2.2 亚甲蓝用量的选择
固定其他的条件,分别取0.50,1.00,1.50,2.00,2.50和3.00 mL亚甲蓝溶液,具体结果见图3。在亚甲蓝用量不断增加的情况下,吸光度也在不断地增加,ΔA先缓慢增加,当亚甲蓝的用量为2.00 mL时,ΔA最大,随后ΔA降低。试验结果表明:亚甲蓝溶液的最适使用量为2.00 mL。
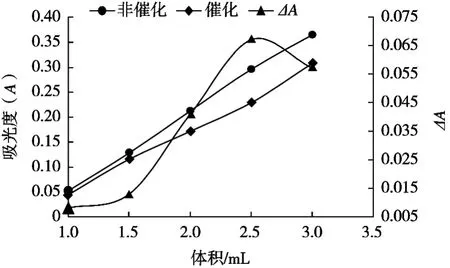
图3 亚甲蓝用量对反应的影响
2.2.3 溴酸钾用量的选择
固定其他的条件,分别吸取1.50,2.00,2.50,3.00,3.50和4.00 mL溴酸钾溶液。具体结果见图4。由图4可知,ΔA的变化随着溴酸钾加入量的增加而增大,溴酸钾在2.50 mL后增长变缓,所以选择溴酸钾用量2.50 mL。
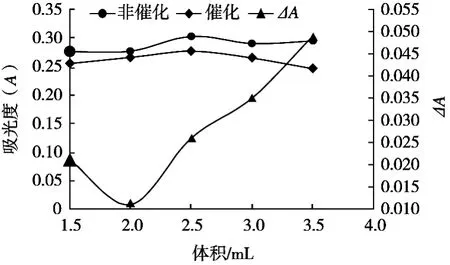
图4 溴酸钾用量对反应的影响
2.2.4 反应温度的选择
反应温度发生改变,吸光度随之发生变化。为了得出温度对体系的影响,将其他条件固定,分别选取不同的温度(65,70,75,80和85 ℃),结果见图5。随着反应温度的不断升高,ΔA也在不断地升高,在75 ℃时达到最大值,试验结果表明:试验在室温下几乎不发生反应,故选择最适反应温度75 ℃。
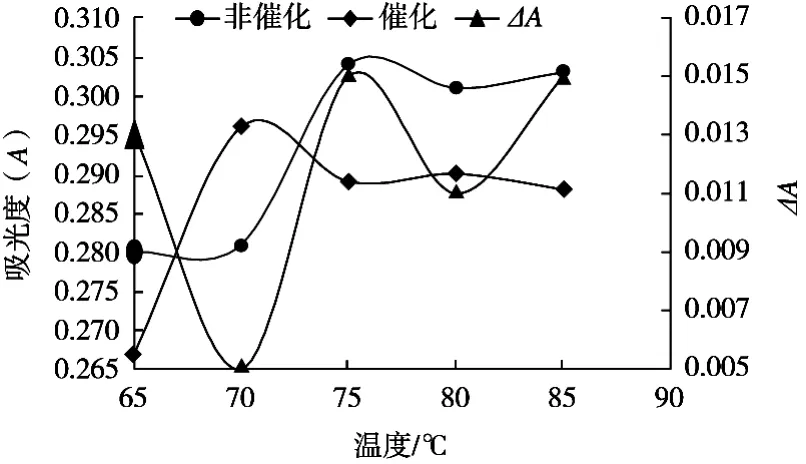
图5 温度对反应的影响
2.2.5 水浴时间的选择
其他条件固定,设定水浴加热时间2,3,4,5,6和7 min,具体结果见图6。由图6可知,ΔA随着反应时间的不同先变化不明显后来又增大而后又减小,此现象发生的原因可能是时间差的选择过小,导致变化不明显。在5~6 min之间,ΔA处于上升期,考虑在5 min时吸光度较大,故选择水浴时间5 min。

图6 水浴时间对反应的影响
2.2.6 工作曲线
在确定了最佳反应条件后,固定其他条件不变,通过改变硒的加入量进行试验,分别取0,1,2,3,4和5 mL的硒标准使用液。由图7可知,工作曲线回归方程为y=5.742 9x-0.002 7,相关系数R2=0.998 6,方法的准确度及灵敏性较好,适用于测定食品中的痕量硒。
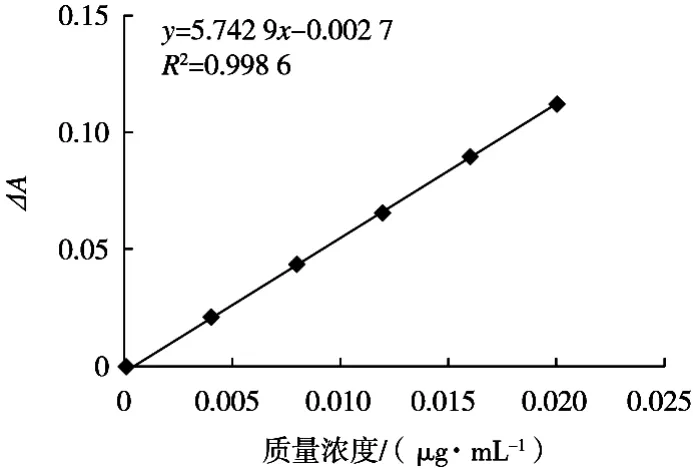
图7 标准曲线
2.3 样品结果分析
按1.4小节所述方法,分别检测黑木耳、核桃、红小豆中硒含量,具体结果见表1~表3。

表1 黑木耳含硒量的结果分析
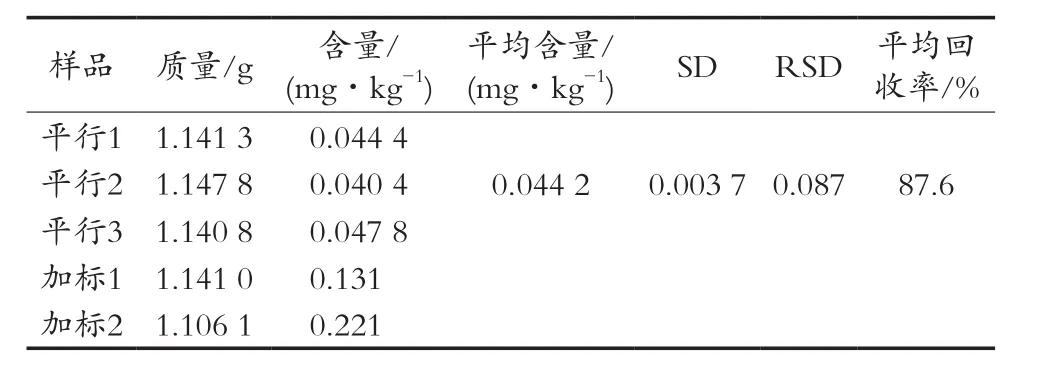
表2 核桃含硒量的结果分析
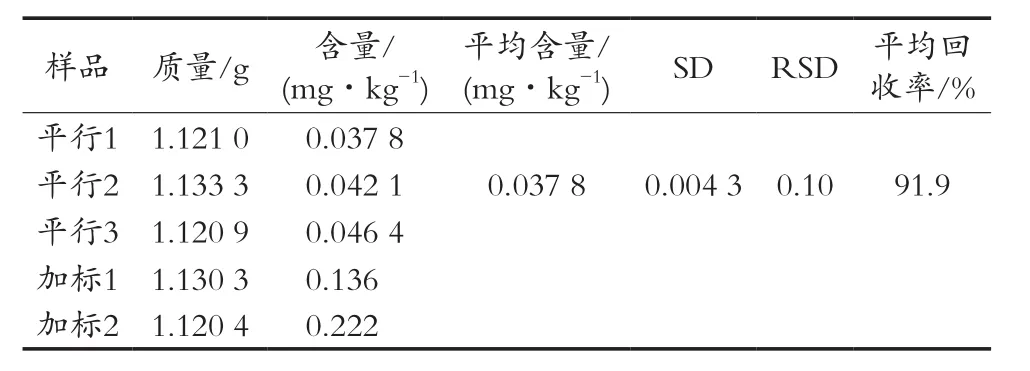
表3 红小豆含硒量的结果分析
通过上述分析可以得出,试验所选用的最适波长为665 nm,稀硝酸的用量为2.00 mL,溴酸钾的用量为2.50 mL,亚甲蓝的用量为2.00 mL,在75 ℃水浴中加热5 min,黑木耳的含硒量为3.78 μg/100 g,核桃的含硒量为4.24 μg/100 g,红小豆的含硒量约为3.76 μg/100 g。测定结果的相对标准偏差在10%左右,回收率在80%~110%之间,符合国标规定,结果较好。
2.4 催化动力学光度法与荧光分光光度法测定结果比较
取样品黑木耳、核桃、红小豆,在相同的消化条件下,分别使用催化动力学光度法和国标GB 5009.93—2017第二法荧光分光光度法,进行6次平行测定,以此来检测方法的精密度,最终测定的相对标准偏差均小于0.1,符合误差允许的范围。对两种方法进行F检验,所得F值均小于F表(p=95%),说明两种方法在精密度上不存在显著性差异。具体结果见表4。
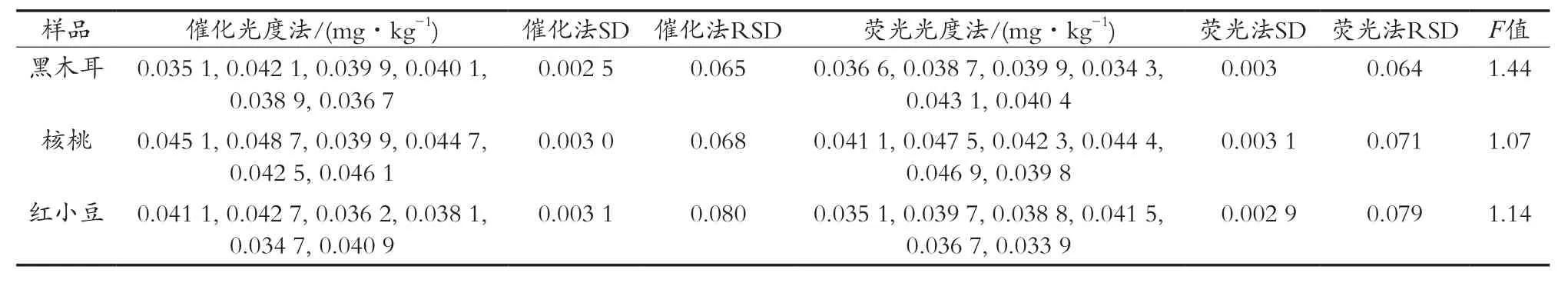
表4 方法的精密度
2.5 测定方法的检出限及定量限
对试剂空白进行11次平行测定,按公式MDL=3×S计算检出限MDL,按公式MQL=10×S计算定量限MQL,式中S为重复测空白中硒11次的标准偏差。结果见表5。

表5 检出限与定量限
3 结论
通过上述分析可得出结论:催化动力学光度法测定食品中硒含量的最适波长为665 nm,稀硝酸的用量为2.00 mL,溴酸钾的用量为2.5 mL,亚甲蓝的用量为2.00 mL,在75 ℃水浴中加热5 min,黑木耳的含硒量为3.78 μg/100 g,核桃的含硒量为4.42 μg/100 g,红小豆的含硒量约为3.78 μg/100 g。加标回收率在80%~110%之间,检出限为0.015 μg/L,方法与国标第二法相比在精密度上不存在显著性差异。与国标中方法相比,方法操作简单,所用仪器价廉易得,准确性较高,成本花费较少。
猜你喜欢
中国典型病例大全(2022年12期)2022-05-13
中国测试(2021年4期)2021-07-16
四川水泥(2018年8期)2018-09-05
食品与生物技术学报(2018年3期)2018-03-25
广东饲料(2016年7期)2016-12-01
健康之家(2016年9期)2016-09-14
广东饲料(2016年8期)2016-02-27
食品界(2016年4期)2016-02-27
现代食品(2015年24期)2015-12-16
现代检验医学杂志(2014年1期)2014-02-06
