
染色体微阵列分析技术在210例发育迟缓/智力低下患儿中的应用
2019-07-16 09:54童光磊鲍劲松李司南周陶成蔡云飞董文旭陈露露康倩倩
安徽医科大学学报 2019年6期
李 红,童光磊,张 敏,鲍劲松,李司南,周陶成,易 昕,蔡云飞,董文旭,陈露露,康倩倩,陈 婧
发育迟缓/智力低下(developmental delay/intellectual disability,DD/ID)是一组在18周岁前发病,以认知功能障碍和社会适应能力缺陷为特征的疾病。一般人群中的发病率较高,为1%~3%[1]。DD/ID的病因较为复杂,主要包括非遗传因素和遗传因素[2]。近年来,国际临床遗传学会(international collaboration for clinical genoemics,ICCG)、国际标准细胞基因组微阵列(international standard for cytogenomic array,ISCA)学会以及美国医学遗传学与基因组学学会(American college of medical genetics and genomics,ACMG)等将染色体微阵列分析(chromosomal microarray analysis,CMA)作为DD/ID患儿的一线临床诊断方法[3-4],染色体微阵列分析是近几年迅速发展起来的一项分子检测技术,能覆盖全基因组DNA,具有高通量、高分辨率、检测速度快的特点。该研究对210例DD/ID患儿进行了CMA检测,以探讨其在诊断、治疗、预后、评估DD/ID患儿方面的价值。
1 材料与方法
1.1 病例资料选取2016年1月1日~2018年8月30日在安徽省儿童医院神经康复科就诊,被确诊为DD/ID的210例患儿临床资料,年龄在2月~10岁。入选标准:① 采用以下测评量表测评:0~4岁使用盖塞尔发育商量表中文修订版(Gessell development scale);4~6岁使用学前和学龄初期中国韦氏儿童智力量表(Chinese Wechsler preschool and primary scale intelligence,WPPSI);>6岁使用中国韦氏儿童智力量表(Chinese Weehsler intelligence scale for children,C-WISC);婴儿初中学生社会生活能力量表;智力商数(intelligence quotient,IQ)/发育商数(developmental quotient,DQ)<70分,同时伴社会适应性能力缺陷者诊断为智力低下。患儿伴或者不伴生长迟缓等其他表现;② 排除颅内出血、中枢神经系统感染、宫内窘迫等影响中枢神经系统发育的相关病史;③ 排除脆性X综合征、苯丙酮尿症、三体综合症等已知遗传因素导致的智力低下。根据是否合并其他异常将DD/ID患儿分成2组,分别为单纯性DD/ID组和合并其他异常组120例(合并癫痫、合并头颅磁共振异常、合并心血管系统、合并多发结构畸形)。所有家长接受检测前遗传咨询及签署知情同意书,该研究获该中心医学伦理委员会批准。
1.2 方法
1.2.1标本采集及处理 采集患儿静脉血2 ml,EDTA抗凝管2~8 ℃保存运输,用于CMV检测。
1.2.2基因组DNA提取 DNA提取试剂盒与提取仪(Lab-Aid820核酸提取仪)购自厦门致善生物科技有限公司;SNParay芯片及扫描仪购自美国Ilumina公司。
1.2.3染色体微阵列检测 严格按照操作说明,应用珀金埃尔默医学检验所用含有60 K寡核苷酸探针的CGX v1.1芯片进行染色体片段重复/缺失检测,应用Iluminaiscan进行芯片扫描、GenomeStudio软件进行数据处理。从软件分析的结果中,选择>100 kb的重复或缺失片段进行下一步分析;对照原始图像检测假阳性和假阴性结果;除外在健康人群拷贝数变异(copynumbervariations,CNVs)多态性的片段;除外片段区域内不包含基因的CNVs。查阅4种常用国际病理性CNV数据库:ClinVar(htp://www.ncbi.nlm.nih.gov/clinvar/)、Decipher(htps://decipher.sanger.ac.uk/)、OMIM(www.ncbi.nlm.nih.gov/omim)和健康人群DGV数据库(database of genomic variants, htps://projects.tcag.ca/variatin),并检查 PubMed数据库相关文献对检出的CNVs致病性进行分析。
1.3 统计学处理应用SPSS 16.0软件进行分析,两组率或多组率的比较采用χ2检验,P<0.05为差异有统计学意义。
2 结果
2.1 CMA结果CMA分析显示210例DD/ID患儿的基因组都存在CNVs,每例患者基因组含1~10个CNVs,片段大小50 kb~50 Mb不等,CMA在83例患儿中检测到染色体拷贝数异常,检出率为39.5%(83/210)。其中,检出已知致病的常见微缺失/微重复综合征66例(79.5%,66/83),见表1;罕见综合征1例(1.2%,1/83,Kleefstra综合征),见图1。可疑致病变异的微缺失/微重复9例(10.8%,9/83),可能良性变异1例(1.2%,1/83),临床意义不明6例(7.2%,6/83)。
2.2 分组及致病性CNVs的检出率临床患儿的表型并不单一,根据伴或不伴其他异常DD/ID患儿分为2组,单纯性DD/ID组90例,复杂性DD/ID组120例(合并癫痫23例,头颅磁共振异常36例,心血管系统19例、多发结构畸形42例)。并对两组检出率进行统计分析(表2)。
3 讨论
CMA能够分析全基因组范围的微缺失或微重复(<5 Mb)、杂合性缺失及单亲二倍体等,目前已成为不明原因发育迟缓、智力障碍、多发先天性畸形等多种疾病的首选检测技术[5]。目前越来越多的研究将CMA作为DD/ID患者的一线检测技术应用于临床诊断中,但所用的平台不尽相同,致病性CNVs检出率也存在差异性。Coutton et al[6]应用Human Genome CGH Microarray Kit 180K技术平台对66例DD患者进行检测,结果发现致病性CNVs的检出率为21%。王荣跃 等[7]应用CytoScan750K对489例DD/ID患儿进行检测,结果显示致病性CNVs的检出率为25.8%。袁海明 等[8]应用CytoScanHD(195万CNV探针+75万SNP探针)芯片对2 000例出生缺陷患儿进行检测,结果发现致病性CNVs的检出率为21.6%。本研究的210例DD/ID患儿结果显示,致病性CNVs的检出率为39.5%,检出率高于国内外文献报道,证明了CMA技术可以弥补传统核型分析技术和FISH技术的不足,提高微小致病性CNVs的检出率。
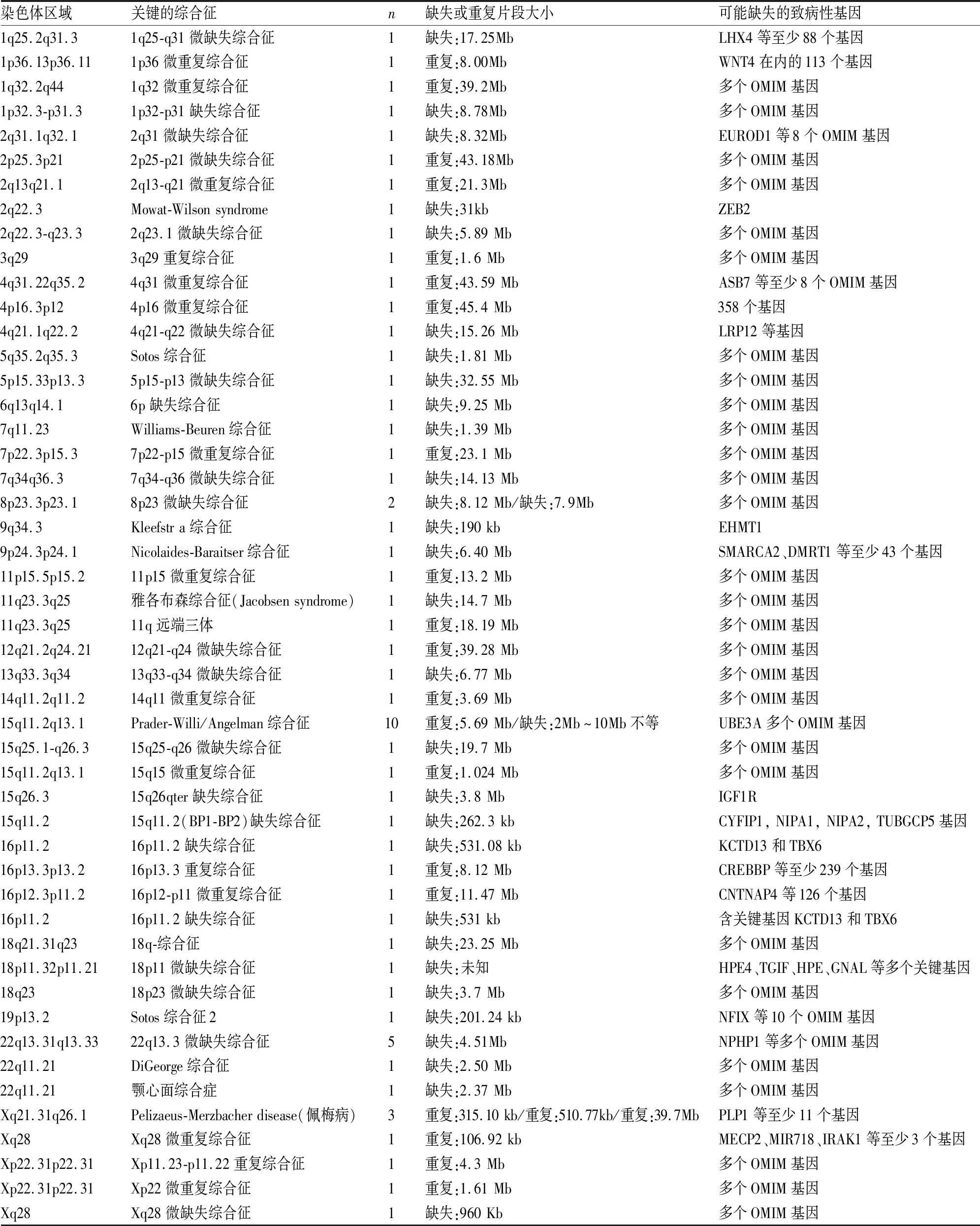
表1 检测出的临床致病性微缺失/微重复综合征在染色体的分布
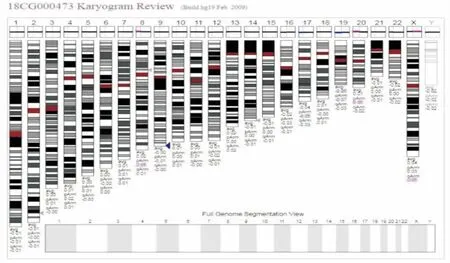
图1 1例罕见微缺失/重复综合征的CMA结果图
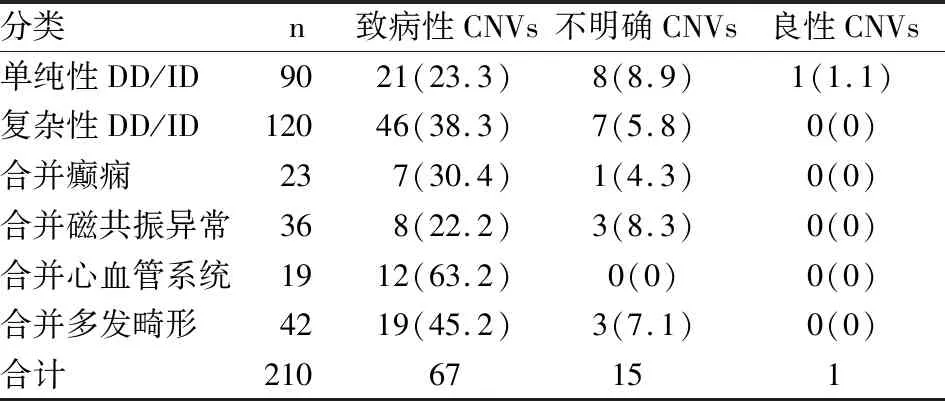
表2 210例患儿分组及致病性CNVs的检出率[n(%)]
研究中67例致病性CNVs,66例(31.4%,66/210)为常见微缺失/微重复综合征,阳性例数大于3例的为Angelman/Prader-Willi综合征10例、22q13.3微缺失综合征5例、佩梅病3例(Pelizaeus-Merzbacher disease,PMD)。Angelman/Prader-Willi综合征多见儿童,表现为快乐表情、发育迟缓、语言障碍、孤独内向、智力儿童低下,癫痫发作;其临床表现与染色体异常具有密切联系,染色体15q11-q13区段的UBE3A基因异常是该病发生的主要因素,不同的突变类型有不同程度的表现,缺失片段的大小与表型具有相关性[9-10]。研究中Angelman/Prader-Willi综合征中6例属于单纯组,4例属于复杂组,说明并不是疾病复杂的发病率就高。PMD致病基因位于Xq22.2的PLPl基因,该基因缺陷可以导致髓鞘形成异常和(或)少突胶质细胞死亡,使得脑内广泛白质区域髓鞘缺乏或减少是累及中枢神经系统白质的进展性遗传性疾病,头颅MRI对本病诊断意义重大,可显示为髓鞘化异常,T2加权像和Flair像弥漫性高信号[11],研究的3例PMD均属于合并磁共振异常组,建议临床医师对发育迟缓,头颅MRI白质异常患儿要排除此病,进行基因芯片检查。
检出Kleefstra综合征1例, Kleefstra综合征是比较罕见的基因组学疾病,由染色体区域9q34.3(85%的病例)中的微缺失或由编码的常染色质组蛋白甲基转移酶1的EHMT1基因突变引起的[12]。本例患儿为12个月女孩,主要表现生长发育迟缓,智力低下,先天性心肌病,特殊面容不明显。CMA在染色体9q34.3区段检出190 Kb缺失,数据库提示为Kleefstra综合征。该综合征患者在国内尚未见报道,不仅丰富了Kleefstra综合征基因型-表型谱,还为治疗和预后评估提供了遗传学依据。
本研究对单纯性DD/ID组与复杂性DD/ID组致病性CNVs检出率比较,差异无统计学意义。说明DD/ID的临床表型具有多样性,没有特异性。
CMA仍具有局限性:① 无法检测染色体的平衡性改变;② 无法检测单基因病,进一步高通量测序可弥补基因突变而导致的漏诊。CMA可检测出核型分析无法识别的微缺失/微重复综合征,还具有发现新的可疑致病性基因的能力,但新发致病基因的确定要结合父母的DNA分析,本研究中不足点是没有开展父母DNA的CMA检测工作。CMA为DD/ID疾病的诊断、咨询、治疗、预后评估以及疾病的再发风险评估提供了遗传学依据。
猜你喜欢
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
中成药(2017年12期)2018-01-19
小天使·二年级语数英综合(2017年4期)2017-04-18
小天使·四年级语数英综合(2017年4期)2017-04-18
恋爱婚姻家庭·养生版(2016年9期)2016-09-07
湖南畜牧兽医(2016年3期)2016-06-05
川北医学院学报(2015年5期)2015-12-05
中央民族大学学报(自然科学版)(2015年2期)2015-06-09
少年文艺·开心阅读作文(2014年5期)2014-10-08
