
苏氨酸醛缩酶的研究进展
2019-07-16 01:00:06黎军张志雄杨金亮李学英
生物技术通讯 2019年3期
黎军,张志雄,杨金亮,李学英
1.遵义医科大学 医学遗传学教研室,贵州 遵义 563000;2.四川大学 生物治疗国家重点实验室,四川 成都 610000
苏氨酸醛缩酶(threonine aldolases,TAs)主要存在于微生物和植物中[1-9],以磷酸吡哆醛(pyridoxal phosphate,PLP)为辅酶,在生理条件下催化苏氨酸裂解为甘氨酸和乙醛,承担苏氨酸分解代谢和甘氨酸合成的功能[8]。在体外,TAs可以催化不同类型的醛与α-氨基酸发生醇醛缩合反应,生成具有2个手性中心的β-羟基-α-氨基酸[10-13],而后者为一类存在于氯霉素、氟苯尼考、甲砜霉素、肾上腺素、屈昔多巴、万古霉素、鞘脂菌素等许多活性医药物成分(active pharmaceutical ingredient,API)中的核心片段[14-19]。因此,TAs在生物合成中具有重要的应用价值。TAs在催化过程中可以严格控制产物α-碳位的立体构型(e.e.>99%),而对β-碳位的立体构型控制相对较弱,产物常为一对非对映异构体[7-12]。在生物合成中如何控制产物βC位的立体构型,以及酶催化过程的分子机制,是近年来研究TAs的热点。
1 TAs的分类
根据其对立体异构的苏氨酸的活性的不同,可将TAs分为L-TAs和D-TAs这2大类。L-TAs可以进一步分为特异性裂解L-苏氨酸的L-TA(EC4.1.2.5)、特异性裂解L-别-苏氨酸的L-allo-TA(EC4.1.2.49)、同时对L-苏氨酸和L-别-苏氨酸均具有裂解活性的低选择性L-low-TA(EC4.1.2.48)等3个类型。D-TAs中,目前仅发现低特异性的 D-TA(EC4.1.2.42)[8,14]。L-TAs和 DTAs发挥作用必须依赖于辅酶PLP,PLP是维生素B6的活性形式。序列相似性和系统发生分析表明L-TAs和D-TAs基因是2个相互独立进化的基因家族。L-TAs属于天冬氨酸氨基转移酶超家族,为PLP依赖的Ⅰ型折叠酶;D-TAs属于丙氨酸消旋酶超家族,为PLP依赖的Ⅲ型折叠酶[8]。
2 TAs的结构
几个种属来源的L-TA、L-low-TA、L-allo-TA、D-TA的晶体结构已解析(表1),为理解TAs的分子催化机制奠定了基础。
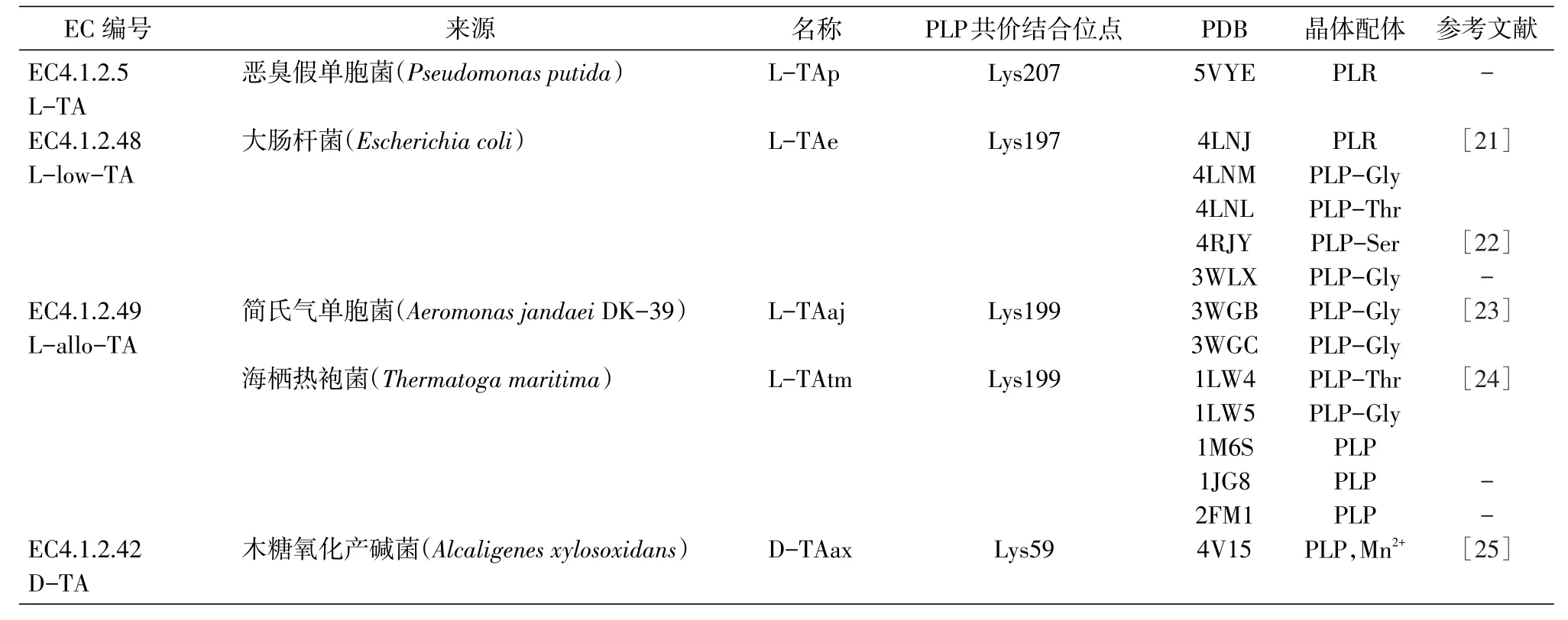
表1 TAs的晶体结构列表
2.1 L-TAs的结构
L-TAs为222对称的同源四聚体,每个单体包含1个大的结构域和1个小的结构域,整体二级结构相似度很高(图1)[21-24]。L-TAs单体大结构域为α/β/α三明治折叠模式,共包含7个β折叠,大结构域位于四聚体中间位置,PLP的醛基共价结合于大结构域的特定赖氨酸残基;L-TAs的小结构域通常由2~4个β折叠和α螺旋构成,位于四聚体的外围。2个单体紧密接触形成催化二聚体,活性位点位于2个单体紧密接触的界面处,每个催化二聚体含有2个活性中心,共有4个活性中心。L-TAs的活性中心具有极其相似的保守氨基酸残基和高度相似的空间结构,PLP的功能基团与这些保守的氨基酸残基形成特定的相互作用(图1C)。
L-TAe(大肠杆菌)的活性中心,PLP与Lys197共价结合形成内部醛亚胺,但在pH5.6条件下PLP与Lys197无法形成内部醛亚胺[22]。PLP的吡啶环与His83的咪唑环形成π堆叠,与Ala168形成疏水接触;吡啶环的氮原子与Asp166的羧基形成氢键相互作用,酚羟基与Arg169的胍基形成氢键相互作用,磷酸基团直接与Arg229的胍基相互作用。活性中心含有2个重要的水分子,一个水分子与PLP的磷酸基团,Ser196、Ser205的羟基,Gly204的酰胺氧形成氢键相互作用;另外一个水分子介导了PLP磷酸基团与Gly227和Lys222形成氢键相互作用[21]。L-TAe可结合6个镁离子或钙离子,其中4个二价阳离子距离活性中心9 Å,另外2个位于四聚体对称中心。
L-TAaj(简氏气单胞菌 DK-39)的 Arg171、Arg313、Ser8的侧链与醛亚胺相互作用,将底物锚定到活性位点,His85的咪唑环与PLP-GLY的吡啶环形成π堆叠,并且可参与调节苏氨酸醛缩酶的立体特异性。配体Gly的氨基略微偏离PLPGly的吡啶环平面,使其易断裂的外部醛亚胺键垂直于PLP-Gly的吡啶环平面。His128的侧链可与底物L-allo-Thr上的羟基形成氢键,当His128突变为Tyr时,残基的侧链从活性位点移出4.2 Å。与野生型酶相比,L-Taj的H128Y/S292R突变体对L-allo-Thr的活性提高了3倍,对LThr的活性提高了322倍。
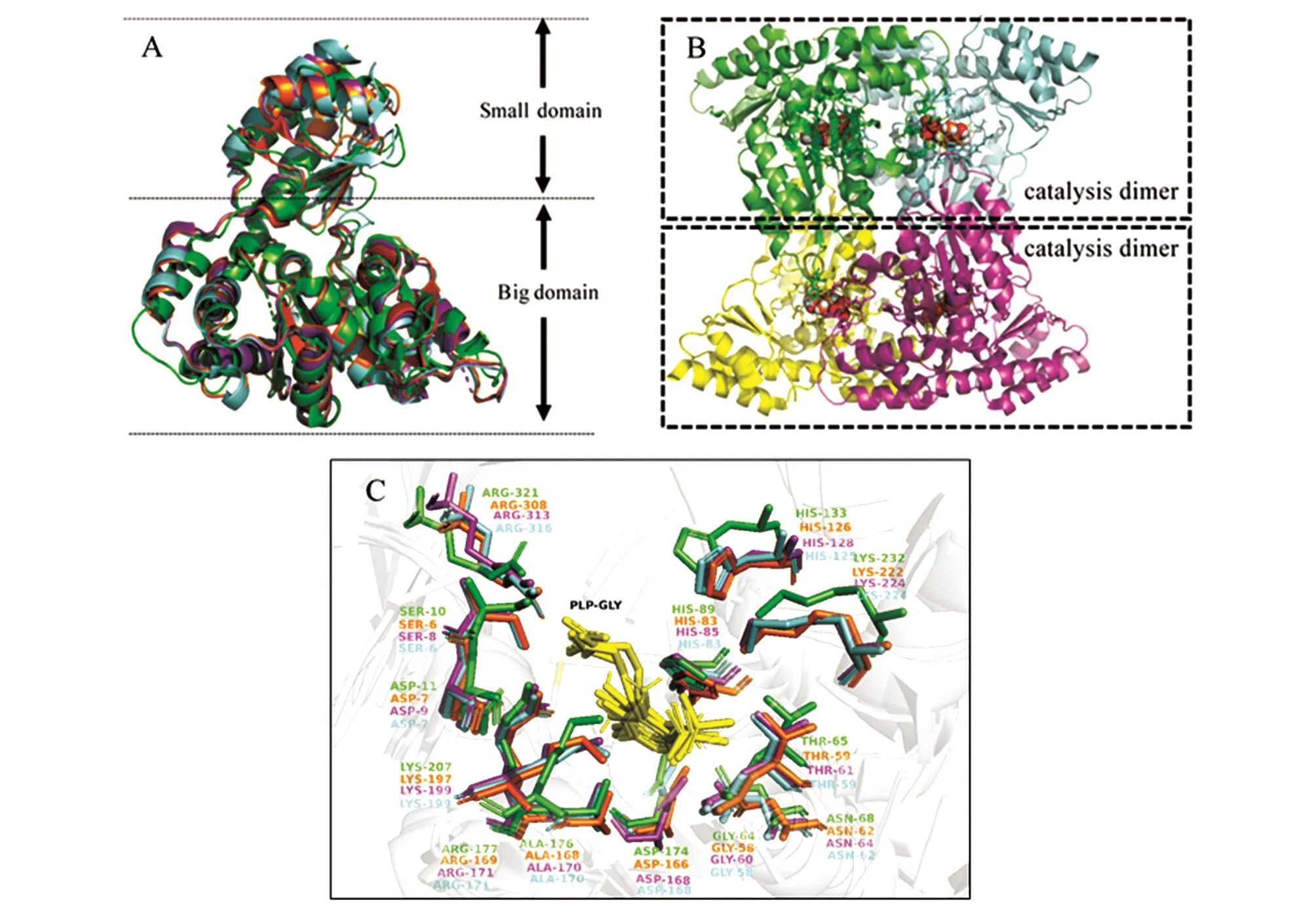
图1 L-TAs的二级结构与活性中心
L-TAtm(海栖热袍菌)与L-TAaj具有相似的整体结构,用DALI服务器(Holm&Sander,1995)进行结构比较,L-TAaj与L-TAtm的晶体结构有49%的一致性(Z-score 47.9;r.m.s.d.1.2 Å)[23]。L-TAtm含有钙离子和钠离子,钙离子位于每个单体的大小结构域之间,四聚体包含6个钙离子,有2个位于四聚体对称中心,每个钙离子通常含有1到2个水分子;钠离子位于Arg72,含有2个水分子。金属离子可能具有稳定四聚体的作用。
2.2 D-TA的结构
D-TAax(木糖氧化产碱菌)为同源二聚体,2个单体头尾相连,接触面共同组成2个活性中心(图2)[25]。每个单体含有2个结构域,大的结构域由8条α/β-桶状结构组成的丙氨酸消旋酶样结构域(32~274残基),小结构域由N端(1~31残基)和C端(275~379残基)组成的β-结构域构成。β-结构域包含5个反相平行的β链组成的β桶装蛋白(275~347残基)和3个β链组成的β片层。活性中心的辅酶PLP与大结构域保守的Lys59可共价结合,PLP 的吡啶环与 Tyr187(~3.7 Å)形成π堆叠。PLP的其余基团主要通过形成氢键与酶结合:PLP的吡啶环的氮原子与Gln249形成氢键;PLP的磷酸基团与Thr233、Ser252和Tyr260的羟基,与Thr233和Gly251的氨基形成氢键;PLP的吡啶环的羟基与Gln81的侧链氨基及Arg157的胍基形成氢键。
D-TAax的催化活性中心含有二价锰离子。锰离子与PLP的醛基相距5距离吡啶环共轭面 0.4 Å,距 离 His347 的 Nε 原 子 2.2 Å,距 离Asp349的羧基2.2 Å。锰离子与4个水分子配位(1.8~2.3 Å)形成一个扭曲的八面体,作为路易斯酸参与D-TAax的催化过程,对D-TA的立体选择性具有重要作用[25]。
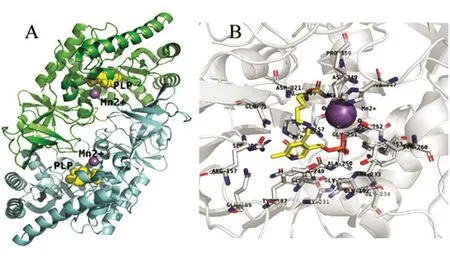
图2 D-TA的结构与活性中心
3 TAs的催化机制
3.1 辅酶PLP介导的催化循环
TAs处于非催化状态时,PLP与活性中心特定的赖氨酸残基形成席夫碱(内部醛亚胺)。含有氨基的底物(Gly、β-羟基-α-氨基酸)进入活性中心,从内部醛亚胺中置换赖氨酸的ε-氨基,与PLP形成新的席夫碱(外部醛亚胺)[14]。外部醛亚胺是TAs催化发生羟醛缩合和逆向羟醛裂解的共同中间体,Gly是所有TAs通用的α-氨基酸供体[9]。在羟醛缩合过程中,Gly与PLP形成外部醛亚胺PLP-Gly,吡啶环帮助α-碳的质子转移至酶催化中心的广义碱,产生高度共振的阴离子;醛受体进攻α-碳形成C-C键,产物通过席夫碱交换机制被释放出活性中心,PLP则从产物转移回到活性中心的保守赖氨酸的侧链重新形成内部醛亚胺,完成一个催化循环。逆向羟醛裂解过程是由外部醛亚胺βC位的羟基去质子化开始,接着进行C-C键的断裂,最终被裂解为醛和Gly[8,20,22,25]。
3.2 TAs的立体选择性机制
Uhl等[25]比较了D-TAax和L-TAtm的活性中心,发现活性中心的位点近似镜像对称[25],可以理解为L-TAtm的活性中心的入口位于PLP吡啶环的si面(si-face),D-TAax的活性中心入口位于PLP的re面(re-face)(图3)。因此,当Gly供体与PLP形成外部醛亚胺后,醛受体只能从辅酶PLP吡啶环的si面进攻α-碳,从而严格地控制L-TAs对α-碳的立体选择性。D-TAs与L-TAs恰好相反的是,醛受体只能从辅酶PLP吡啶环的re面进攻外部醛亚胺的α-碳,也能非常严格地控制DTAs对α-碳的立体选择性。
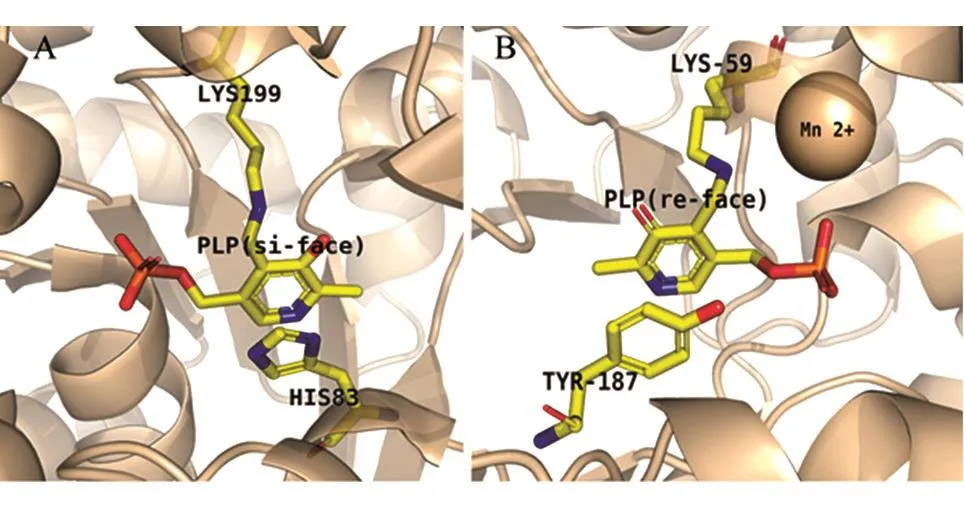
图3 L-TA与D-TA的PLP结合构象呈镜面对称
Fesko等[14]进行了一项核磁共振研究,以了解为何L-TAs催化产物β-碳位的快速差向异构化而D-TAs没有进行快速差向异构化的化学机制。基于PLP介导的TAs催化循环机理,可以推导出酶促反应过程中产生的中间体(外部醛亚胺PLP-Gly)和底物(Gly、醛受体),将甘氨酸、苯甲醛、酶,以及13C标记的顺式产物(syn)与未标记的反式(anti)产物以60∶40的比例混合,测定从顺式产物到反式产物和游离Gly的13C标记转移的相对速率。在动力学控制反应中,L-TA具有中等选择性,D-TA具有高的选择性,顺式产物为主要部分,而对于L-allo-TA和L-low-TA的产物主要为反式产物。在热力学控制下,对于所有TAs,得到60/40(syn/anti,d.e.≈20%)的比例。L-TA的限速步骤是甘氨酸与酶活性位点的结合形成Gly-PLP复合物,以及α-碳去质子化的过程,需要较高的活化能。D-TAs的限速步骤是C-C键的形成,在动力学控制下D-TA可以获得高非对映选择性产物(d.e.>95%,syn)[14]。
4 TAs的定向进化
天然的L-TAs无法满足工业应用的需求。酶的定向进化是提高酶的催化效率、立体选择性、热稳定性的有效手段。目前,L-TAs定向进化策略主要包括两类,一类是利用L-TAs基因缺陷型工程菌直接进行筛选[20],一类是通过检测L-TAs的催化裂解产物乙醛或芳香乙醛进行筛选[26]。
基因工程改造大肠杆菌使其Gly营养缺陷,含有外源L-TAs基因的阳性克隆可以裂解手性底物产生Gly来维持正常生长,这种方法可用于以Gly为供体的催化反应[27]。利用恶臭假单胞菌KT2440可以苯甲醛作为碳源进行生长的特点,首先使其失去内源性的TAs,通过质粒导入待筛选的TAs基因。阳性克隆以手性苯基丝氨酸作为底物,产生可供其利用的苯甲醛,维持正常增殖[20],这种方法适用于筛选以芳香醛为醛受体的催化反应。TAs裂解目标底物产生乙醛或芳香醛,利用乙醛脱氢酶(ALDH)在乙醛存在条件下还原NADH,直接测定340 nm波长下的光密度变化间接检测TAs的催化活性[28]。采用乙酰丙酮分光光度法测定乙醛,也适用于高通量筛选[26]。
Baik等对来源于天蓝色链霉菌(Streptomyces coelicolor)的L-low-TA进行定向进化,使其催化合成L-threo-DOPS(屈昔多巴)的d.e.值从14%(syn)提升到55%(syn)[29]。第1轮筛选采用自动化的克隆筛选系统(QARRAY lite,X2601,Genetix),挑选了25 000个克隆,在深孔板中培养后,以L-苏氨酸为底物,利用乙酰丙酮分光光度法检测裂解产物乙醛进行高通量筛选。4个突变体T2-2(V86I/R241C/Y306C)、T2-4(Y34C)、T3-2(R241C/A287V)和T3-3(Y39C,Y306C)的催化活性和立体选择性均较好,非对映异构体产物的d.e.分别为21%(syn)、26%(syn)、21%(syn)和38%(syn)。采用易错PCR技术对T3-3进行随机突变,在20 000个克隆中获得2个立体选择性较高的突变体T3-3m5(Y39C/Y306C/A48T)和 T3-3m7(Y39C,Y306C,R316C),非对映异构体产物的d.e.分别为43%(syn)和28%(syn)。通过2轮筛选发现突变体 T3-3(Y39C,Y306C)、T3-3m5(Y39C/Y306C/A48T)、T3-3m7(Y39C,Y306C,R316C)均含有Y39C和Y306C突变,推测Y39C和Y306C对提高立体选择性具有重要贡献。把T2-4(Y34C)的突变分别引入T3-3、T3-3m5、T3-3m7中,获得新的突变体T3-3mm1、T3-3mm2、T3-3mm3,产物的d.e.分别为52%、55.4%和49%。突变体T3-3mm2以L-threo-DOPS为底物的特异性常数比值为1.8×107,与野生型的特异性常数比值1.1×108相近[17,26,29]。
5 TAs在生物催化合成中的应用
β-羟基氨基酰胺酒石酸是重要的候选药物,其关键中间体(2R,3S)-2-amino-3-hydroxy-3-pyridin-4-yl)-propanoic acid的化学合成须多步反应,且起始物料异腈基乙酸乙酯不稳定、有毒,产物为消旋体[30]。采用D-TAax(木糖氧化产碱菌IFO12699)和 D-TAar(节杆菌DK-38)的催化工艺不仅实现了工艺简化,还降低了成本。二价金属离子钴离子或镍离子可维持重组D-TAax和DTAar在4℃条件下1周内保持酶活性不变。通过工艺优化,从小试放大到100 L体系,获得的产物均具有很高的d.e.(99.1%,syn)[31]。
L-TAs可以直接催化合成氯霉素的中间体L-4-硝基苯基丝氨酸[16]。余进海等[18]研究了大肠杆菌苏氨酸醛缩酶催化合成L-4-硝基苯基丝氨酸的工艺,重点考察了温度、pH、底物浓度、底物比例、反应时间对催化合成的影响。在45℃、pH8.0、甘氨酸浓度500 mmol/L、对硝基苯甲醛浓度100 mmol/L的条件下,反应24 h,转化率为43%,顺式产物与反式产物的比例为2∶1。
L-threo-DOPS为去甲肾上腺素的前体分子,是一种已上市的帕金森症治疗药物。Gwon等以大肠杆菌JM109为宿主菌表达筛选突变体T3-3mm3(Y34C/Y39C/Y306C/R316C),含有该酶的菌体在-80℃冷冻过夜后直接用于催化合成L-threo-DOPS,在最优催化条件(2 mol/L甘氨酸,145 mmol/L 3,4-dihydroxybenzaldehyde,0.6 g/L PLP,50 mmol/L亚硫酸钠,0.0075 g/L Triton X-100,pH6.5)下,产物的d.e.(60%,syn)相比优化前的d.e.(49%,syn)提高了11%[29]。
6 TAs研究展望
D-TA通过工艺优化和动力学控制,可以很好地控制β-碳位的立体选择性,已成功应用于β-羟基氨基酰胺酒石酸的合成。L-TAs还需要进一步的催化机制研究和酶工程改造。通过解析L-TAs与不同立体构型的目标产物或醛受体的复合物晶体结构,有望找到其对β-碳手性控制的关键活性位点。在结构的基础上,可以采用基因突变、计算机模拟(分子动力学、QM/MM等)等研究L-TAs的手性控制机制,寻找潜在的手性控制方法。通过酶工程降低PLP-Gly形成的活化能,亦有望增加L-TAs催化反应的立体选择性。定向进化可以提高TAs的非对映选择性。需要注意的是,在筛选和测试酶活性的策略上,以通用底物苏氨酸为基础的简单活性测试方法可能与具体应用存在较大差异。很有必要直接以目标来建立高通量筛选方法。
开发以L-TAs为生物催化剂的新工艺来生产氯霉素、氟苯尼考、甲砜霉素、肾上腺素、屈昔多巴等原料药的关键技术,不仅有望提高此类原料药的品质,降低合成工艺复杂度,还可以降低生产成本、减少环境污染。
猜你喜欢
生物化学与生物物理进展(2022年6期)2022-07-21 11:52:06
云南化工(2021年10期)2021-12-21 07:33:28
中学生数理化(高中版.高考理化)(2021年2期)2021-03-19 08:52:38
化工学报(2020年4期)2020-05-28 09:25:24
今日农业(2019年11期)2019-08-13 00:49:02
饲料工业(2017年8期)2017-04-05 04:43:34
广东饲料(2016年1期)2016-12-01 03:43:01
池州学院学报(2015年3期)2016-01-05 01:13:04
天津科技大学学报(2015年2期)2015-08-09 01:40:42
饲料博览(2015年4期)2015-04-05 10:34:14
