
利用CRISPR/Cas9技术产生肌肉特异表达Cas9的小鼠胚胎
2019-06-27 06:07:14李昊陈胜男李松美朴善花安铁洙王春生
中国实验动物学报 2019年3期
李昊,陈胜男,李松美,朴善花,安铁洙,王春生
(东北林业大学生命科学学院 动物发育研究室,哈尔滨 150040)
肌肉生长抑制素(myostatin,MSTN),是一种骨骼肌细胞自分泌蛋白,对肌肉生长发育具有负调控作用[1]。MSTN基因过量表达可导致动物或人肌肉萎缩,而MSNT基因突变或敲除可使动物肌肉量增加[2-4]。不过有研究表明,MSNT基因不仅在骨骼肌中特异性表达,在胚胎、乳腺[5]、成体心脏、脂肪、脑[6]、肝[7]、肺、肾、舌、小肠以及骨髓中也检测到MSTN mRNA存在。敲除MSNT基因后,虽然可以提高肌肉量的增加,但同时也存在影响个体发育甚至是致死等问题。对MSTN基因的研究,不仅可以获得具有优良性状的禽畜,也可在医学上为肌肉萎缩症及有关疾病的改善和治疗提供可能性。因此,如何避免MSTN基因研究中的副作用成为目前研究重点。
CRISPR/Cas9系统主要由Cas9核酸酶和sgRNA构成,其中,sgRNA包含一段与靶点PAM序列(NGG)上游互补的序列,可引导Cas9结合到靶位点并发挥作用。基因编辑过程是通过内源性的DNA双链断裂途径,非同源末端连接(NHEJ)或同源定向修复(HDR)来实现。相比传统技术,CRISPR/Cas9具有设计操作简单、成本低、特异性高和打靶效率高等优点,因此成为当今热门研究手段之一,靶向基因编辑已成为一种主流研究方法,在基因改造和构建转基因动物研究中被广泛使用。近几年,不断有利用CRISPR/Cas9技术产生基因编辑动物的报道[8-10]。
为了建立肌肉特异表达Cas9小鼠动物模型,进而研究成体水平上肌肉组织中MSTN的基因编辑。本研究,选取Rosa26作为编辑和打靶位点,通过显微注射构建了Rosa26位点基因编辑小鼠,同时获得了肌肉特异性表达Cas9蛋白的小鼠打靶胚胎,为后续产生肌肉特异表达Cas9蛋白小鼠动物模型奠定基础。
1 材料与方法
1.1 材料
1.1.1 实验动物、菌株和载体
6~8周龄SPF级雌性ICR小鼠200只,体重25~30 g,购于辽宁长生生物技术股份有限公司【SCXK(辽)2015-0001】;8周龄SPF级雄性B6D2F1小鼠10只,体重30~35 g,购于北京维通利华实验动物技术有限公司【SCXK(京)2016-0006】。饲养于东北林业大学生命科学学院SPF实验动物室【SYXK(黑)2015002】。饲养环境:昼夜各12 h交替,温度22~25℃,湿度35%~45%。饲养期间给予小鼠标准饲料及洁净饮用水。所有操作均符合实验动物伦理学要求。
大肠杆菌DH5α感受态细胞购于北京全式金生物技术有限公司。pMD19-Simple载体购于宝生物工程(大连)有限公司,pMD19-SP和px459载体为实验室自存载体。
1.1.2 实验试剂
Endo-free Plasmid Mini Kit II(Omega,D6950-01),Gel Extraction Kit(CWBIO,CW2302M),GeneJET Genomic DNA Purification Kit(Thermo,K0721),TranscriptAid T7 High Yield Transcription Kit(Thermo,K0441),TrueCut Cas9 Protein v2(Invitrogen,A36497),QuickExtract(Lucigen,QE0905T),限制性内切酶(Promega),Taq酶(TAKARA),DNA分子量标记Marker(TianGen),其他试剂如无特殊说明均购于Sigma公司。
1.1.3 实验仪器
梯度PCR仪(Bio-Rad,美国),分光光度计(Thermo,NanoDrop 2000,美国),拉针仪(Narishige,PN-30,日本),锻熔仪(Narishige,MF-900,日本),显微操作仪(Eppendorf,TransferMan NK2,德国),倒置荧光显微镜(Carl Zeiss,Axiovert 200,德国),CCD成像系统(Nikon,DS-Fi1,日本)。
1.2 方法
1.2.1 小鼠Rosa26基因sgRNA的设计与获得
(1)sgRNA的设计
设计Rosa26位点sgRNA,上游引物mRosa26-gRNA-PCRF,下游引物mRosa26-gRNA-universal-PCRR,引物序列详情见表1,引物送哈尔滨睿博兴科生物技术有限公司公司合成。
(2)退火形成双链DNA
反应组成:mRosa26-gRNA-PCRF(10 μmol/L)2.5 μL, mRosa26-gRNA-universal-PCRR(10 μmol/L)2.5 μL,2×Premix LA Taq 25 μL,ddH2O 20 μL,反应体系:50 μL。退火条件:98℃ 2 min;98℃ 10 s、53℃ 20 s、72℃ 30 s、20个循环;72℃ 5 min。
(3)sgRNA的体外转录
反应组成:双链DNA 5 μL,5× reaction buffer 4 μL,GTP solution 2 μL,UTP solution 2 μL,CTP solution 2 μL,T7 RNA polymerase mix 2 μL,补加RNase-free H2O到20 μL,反应体系:20 μL。混匀后,37℃孵育6 h,然后加DNase Ⅰ 1 μL,37℃孵育15 min;纯化,分装,-80℃保存。
1.2.2 sgRNA和Cas9蛋白的体外酶切验证
(1)小鼠Rosa26基因靶DNA的扩增
引物为上游mRosa26-Xba-F1和下游mRosa26-Xba-R1,引物序列详情见表1。反应组成:基因组DNA 1 μL(50 ng),mRosa26-Xba-F1 0.5 μL,mRosa26-Xba-R1 0.5 μL,2×LA Mix 10 μL,补加ddH2O 到20 μL,反应体系:20 μL。扩增条件:98℃ 5 min;98℃ 30 s、58℃ 30 s、72℃ 30 s、35个循环;72℃ 7 min。
(2)sgRNA和Cas9蛋白的体外酶切
将80 ng的Rosa26靶DNA模板和150 ng的Cas9蛋白混合,37℃孵育30 min,加200 ng sgRNA,1 μL NEB Buffer 3和1 μL 10 mmol/L BSA,混合均匀,37℃下作用1.5 h,酶切后进行琼脂糖凝胶电泳,验证酶切效果。
1.2.3 小鼠Rosa26位点肌肉特异性同源打靶载体的构建
(1)SP-px459载体构建
采用限制性内切酶KpnI和AgeI对pMD19-SP和px459载体进行酶切,将回收的SP启动子和去除CMV启动子的px459载体大片段连接,构建SP-px459载体。
(2)SP-px459载体的PCR扩增
设计引物T-MluI-SP-F和T-BstBI-R,并在上下游引物两端引入与Donor载体酶切位点两侧同源的15 bp序列,引物序列详情见表1。反应组成:SP-px459 3 μL、T-MluI-SP-F 3 μL、T-BstBI-R 3 μL、2× Premix LA Taq 50 μL、ddH2O 41 μL,反应体系:100 μL。反应条件:98℃ 1 min;98℃ 10 s、65℃ 30 s、72℃ 7 min、30个循环;72℃ 5 min。
(3)同源臂载体的酶切
使用限制性内切酶MluI和BstBI对小鼠ROSA26 Safe Harbor 基因敲入试剂盒中的DC-DON-SH02 ROSA26(Donor)载体,进行酶切,胶回收大片段,并置于-20℃备用。
(4)Donor-SP-px459载体的构建
取上述PCR胶回收产物40 ng,Donor酶切回收产物50 ng,加1 μL Exnase II,2 μL的5 × CE II Buffer,补加ddH2O到10 μL,进行同源重组连接。
1.2.4Rosa26位点基因编辑小鼠胚胎的获得
选择6~8周大性成熟的雌性ICR小鼠,间隔48 h分别注射10 IU的PMSG和hCG,并与雄性B6D2F1小鼠合笼,见栓小鼠采用输卵管灌流收集原核期胚胎,随后采用原核注射方式将Rosa26 sgRNA和Cas9蛋白注射到受精卵的雄原核,随后观察胚胎的卵裂和囊胚发育情况。
1.2.5Rosa26位点基因编辑小鼠胚胎的鉴定
(1)囊胚收集处理
收集注射后囊胚加入快速DNA提取试剂,每10 μL裂解液放入1枚囊胚。随后进行变性处理,变性程序:68℃ 6 min、98℃ 2 min。
(2)两轮PCR扩增
以变性后的DNA作为模板,以mRosa26-Xba-F1和mRosa26-Xba-R1为引物,进行两轮PCR。一轮PCR反应组成:胚胎裂解液10 μL,mRosa26-Xba-F1 0.3 μL,mRosa26-Xba-R1 0.3 μL,2×Premix LA Taq 10 μL,反应体系:20.6 μL。二轮PCR反应组成:一轮PCR产物5 μL,mRosa26-Xba-F1 0.3 μL,mRosa26-Xba-R1 0.3 μL,2×Premix LA Taq 10 μL,ddH2O 4.4 μL,反应体系:20 μL。PCR 反应条件为:98℃ 3 min;95℃ 30 s、58℃ 30 s、72℃ 45 s、30个循环;72℃ 7 min。
1.2.6Rosa26位点基因编辑小鼠的制作
原核注射后,将发育到囊胚阶段的小鼠胚胎,以每侧7~8枚的数量,通过子宫移植的方法,移植到同步发情的3.5 d或2.5 d受体母鼠子宫,待其生产。
1.2.7Rosa26位点基因编辑小鼠的鉴定
取出生4周小鼠尾尖,提取小鼠尾尖DNA,以mRosa26-Xba-F1和mRosa26-Xba-R1为引物进行PCR扩增,反应组成和扩增条件见1.2.2。PCR产物送生物公司进行测序。
1.2.8Rosa26位点肌肉特异性打靶胚胎的获得
采用1.2.4方法收集小鼠原核期胚胎,同样采用原核注射方式将同源打靶载体Donor-SP-px459以质粒的形式与Rosa26 sgRNA和Cas9蛋白一起注射到受精卵的雄原核中,随后观察胚胎的卵裂和囊胚发育情况。
1.2.9Rosa26位点肌肉特异性打靶胚胎的鉴定
使用小鼠ROSA26 Safe Harbor 基因敲入试剂盒提供的通用鉴定引物3’-Poly(A)-F和3’-Poly(A)-R对打靶载体的整合情况进行鉴定,引物序列详情见表1。两轮PCR反应组成和反应条件同1.2.5。
表1 引物序列Table 1 Primer sequences
2 结果
2.1 靶DNA扩增和sgRNA体外酶切验证
扩增的小鼠Rosa26基因靶DNA大小约为600 bp,通过琼脂糖凝胶电泳,在600 bp左右可以观察到条带,说明扩增成功(图1A)。sgRNA和Cas9蛋白对靶DNA酶切后产生的条带大小约200 bp和400 bp,通过琼脂糖凝胶电泳,结果显示,在体外酶切验证体系中同时加入sgRNA和Cas9蛋白,电泳后可在约200 bp和400 bp处观察到条带,说明酶切成功(图1B),而未加入sgRNA和Cas9或单独加入sgRNA或Cas9蛋白时未观察到特定条带出现。
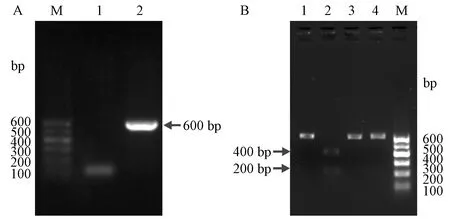
注:A,Rosa26靶DNA PCR产物琼脂糖凝胶电泳图;M,Marker I DNA ladder;1,阴性对照;2,PCR产物。B,Rosa26靶DNA体外酶切验证琼脂糖凝胶电泳图;1,阴性对照;2,sgRNA+Cas9;3,sgRNA;4,Cas9;M,Marker I DNA ladder.图1 Rosa26靶DNA PCR产物和sgRNA体外酶切验证琼脂糖凝胶电泳图Note. A. Agarose gel electrophoresis result of Rosa26 target DNA PCR product. M, Marker I DNA ladder. 1, Control. 2, PCR product. B. Agarose gel electrophoresis results of sgRNA in vitro cleavage assay products. 1, Control. 2, sgRNA + Cas9.3, sgRNA. 4, Cas9.M, Marker I DNA ladder.Figure 1 Results of agarose gel electrophoresis of Rosa26 target DNA PCR and sgRNA in vitro cleavage assay products
2.2 肌肉特异表达同源打靶载体的构建
构建的SP-px459载体的PCR产物大小约为5000 bp,通过琼脂糖凝胶电泳,可以在约5000 bp左右观察到条带出现,说明载体构建成功(图2A)。构建的同源打靶载体Donor-SP-px459的大小约为13 000 bp左右,琼脂糖凝胶电泳后,在约13 000 bp处观察到条带,说明载体构建成功(图2B)。
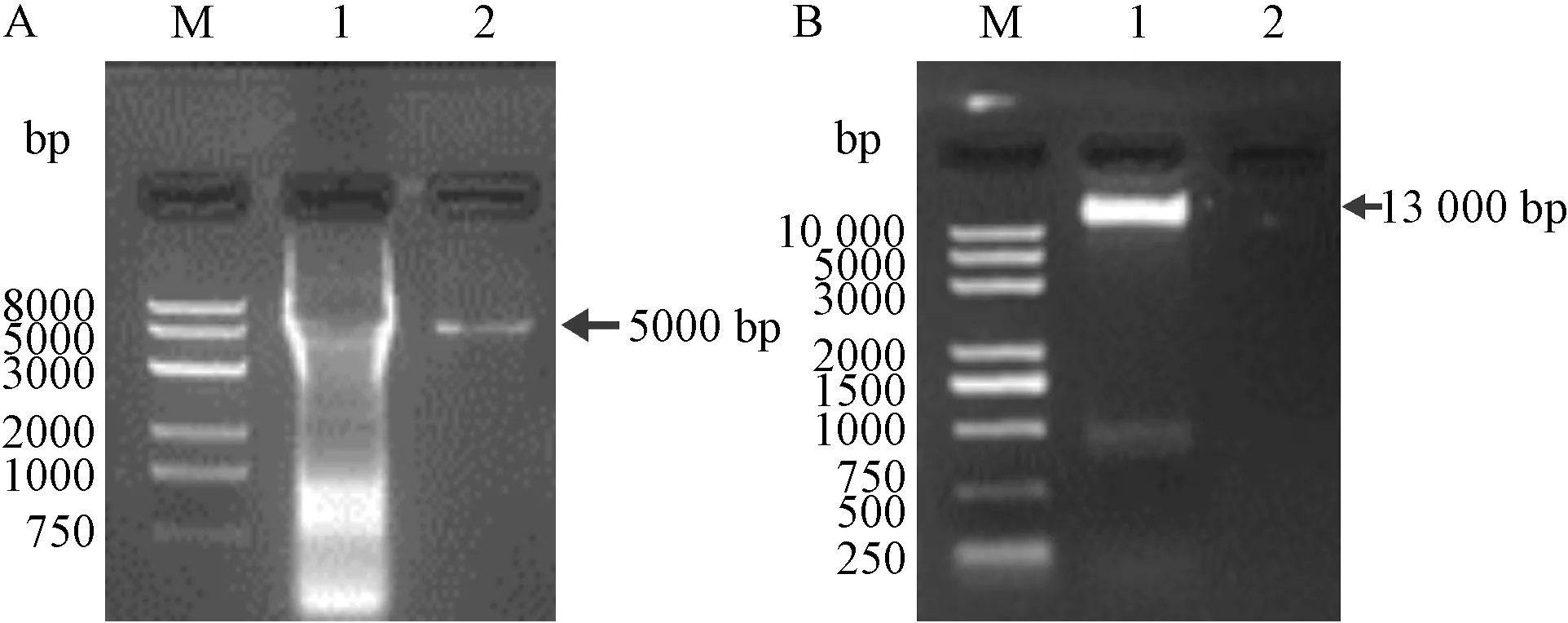
注:A,SP-px459的PCR产物;M,DL 5000 Plus marker;1,阴性对照;2,PCR产物。 B,Donor-SP-px459载体的琼脂糖凝胶电泳图;M,DL 2503 marker;1,Donor-SP-px459载体;2,阴性对照。图2 SP-px459 PCR产物和Donor-SP-px459的载体构建Note. A. PCR products of SP-px459. M, DL 5000 Plus. 1, Control. 2, PCR Products. B. Agarose gel electrophoresis of donor-SP-px459 vector. M, DL 2503 marker. 1, Donor-SP-px459 vector. 2, Control.Figure 2 PCR products of SP-px459 and vector construction of donor-SP-px459
2.3 小鼠Rosa26基因编辑胚胎的获得
注射了Rosa26的sgRNA和Cas9蛋白的胚胎可以正常卵裂并可发育到囊胚阶段(图3)。在挑取了94个原核时期胚胎进行注射后,有85个胚胎存活,有9枚胚胎在注射后发生崩解;注射后观察细胞卵裂情况,其中有78枚胚胎分裂形成二细胞,卵裂率为91.8%;分裂后的胚胎最终由40枚发育到囊胚,囊胚率为51.3%。注射后得到的囊胚(图4A),经PCR扩增,电泳后,20个囊胚中有19个出现了特异性扩增条带(图4B);测序结果显示,在测序的6个样品中有1个发生了基因缺失编辑(图4C,4D)。
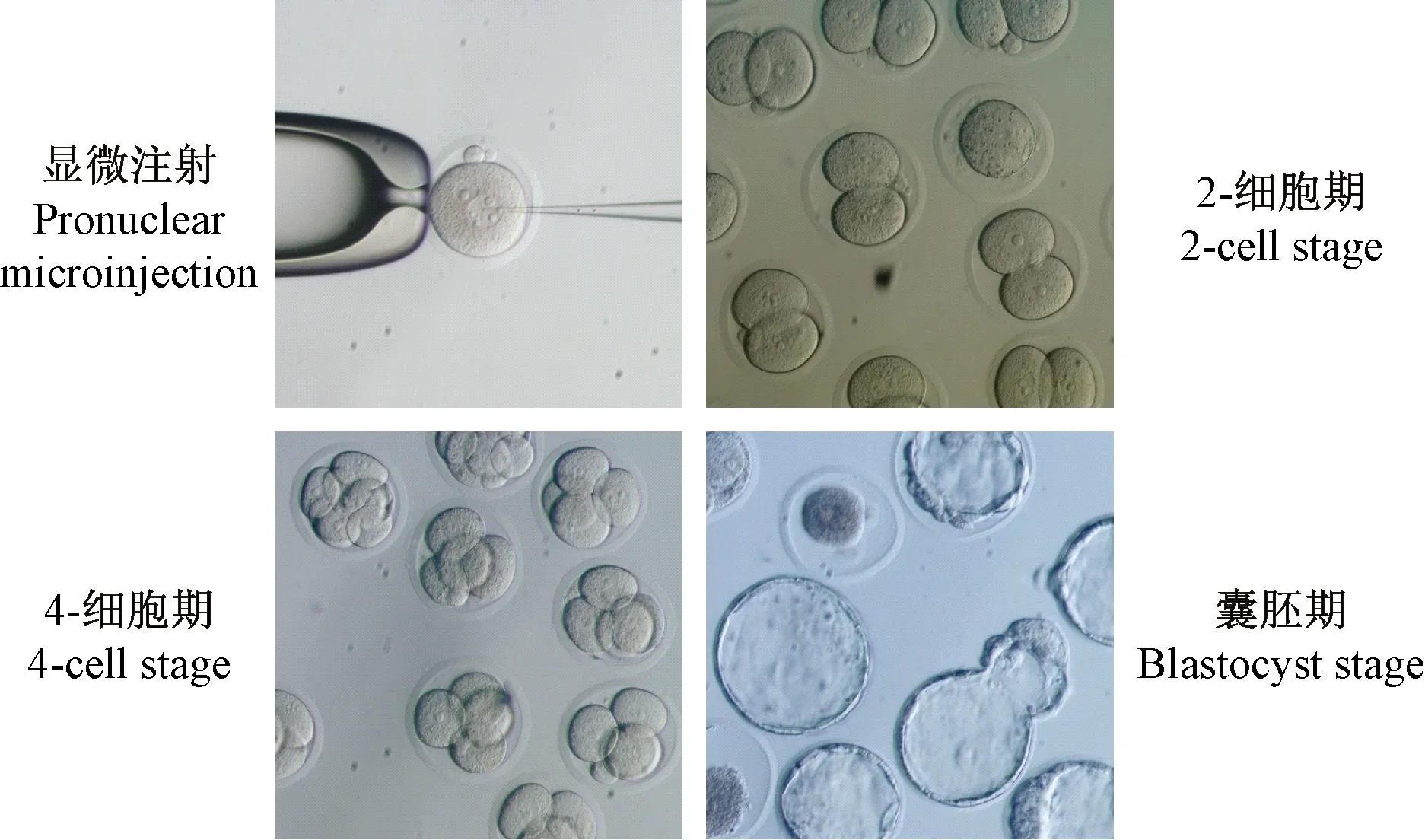
图3 注射Rosa26 sgRNA/Cas9后小鼠胚胎发育过程(×200)Figure 3 Process of embryos development after Rosa26 sgRNA/Cas9 microinjection(×200)
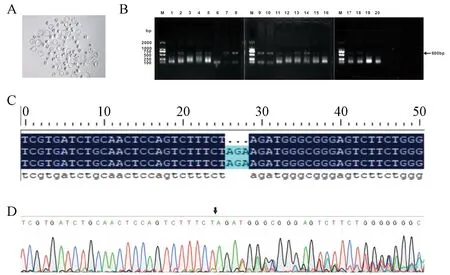
注:A,合子注射后形成的囊胚。B,囊胚PCR产物。C,序列比对结果。D,测序峰图结果。图4 小鼠Rosa26基因编辑胚胎的鉴定Note. A, Blastocysts formed after zygotic injection. B, PCR products of blastulae. C, Sequence alignment. D, Sequencing peak map.Figure 4 Identification of mouse Rosa26 gene edited embryos
2.4 Rosa26基因编辑小鼠的获得
采用子宫移植的方法,将96枚注射后的囊胚移植到同步发情的6只受体体内,其中4只没有观察到妊娠反应,最终有1只产下3只小鼠(图5A)。鼠尾DNA,PCR扩增后经琼脂糖凝胶电泳检测,在600 bp左右出现目的条带(图5B),测序结果显示,其中一只小鼠的Rosa26位点发生了点突变(图5C,5D)。
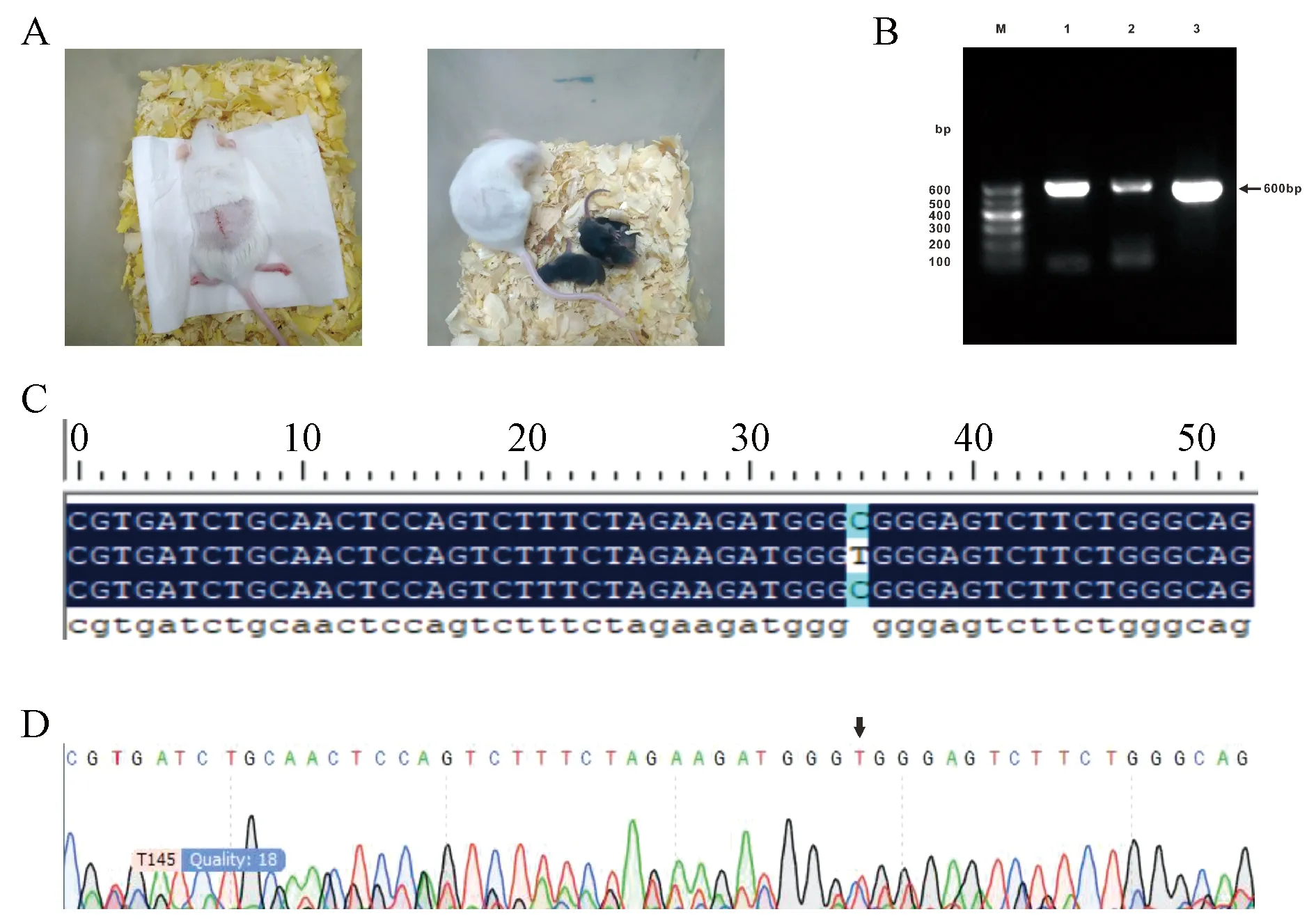
注:A,胚胎移植和获得的后代。B,基因组PCR产物电泳图;1~3:移植后产生的后代。C,序列比对结果。D,测序峰图结果。图5 Rosa26基因编辑小鼠的鉴定Note. A, Embryo transfer and acquired offsprings. B, Electropherogram of genomic PCR identification products, lane1~3, offsprings after embryo transfer. C, Sequence alignment. D, Sequencing peak map.Figure 5 Identification of Rosa26 gene edited mice
2.5 肌肉特异表达Cas9蛋白小鼠胚胎的获得
打靶载体Donor-SP-px459与Rosa26 sgRNA和Cas9蛋白联合注射后,胚胎可以正常卵裂并可发育到囊胚阶段(图6)。注射打靶载体Donor-SP-px459的胚胎的卵裂率为87.1%,囊胚率为27.8%。注射了Donor-SP-px459的胚胎中未见有荧光(图7)。注射了打靶载体的胚胎,PCR扩增后,经琼脂糖凝胶电泳检测,在约1400 bp大小处扩增出特异性条带(图8),说明同源打靶载体已成功整合到打靶位点。
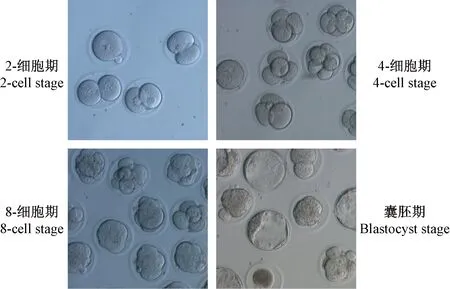
图6 注射打靶载体Donor-SP-px459后小鼠胚胎发育过程(×200)Figure 6 Process of embryos development after Donor-SP-px459 microinjection(×200)
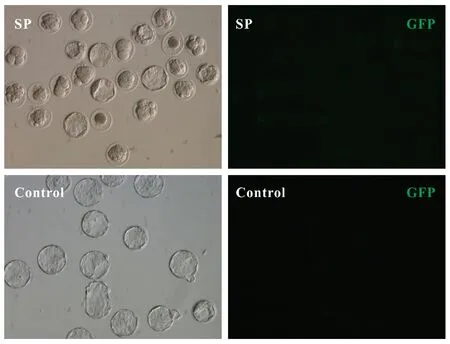
图7 注射打靶载体后胚胎荧光蛋白表达情况Figure 7 Expression of fluorescent protein after microinjection of targeting vector into the embryos
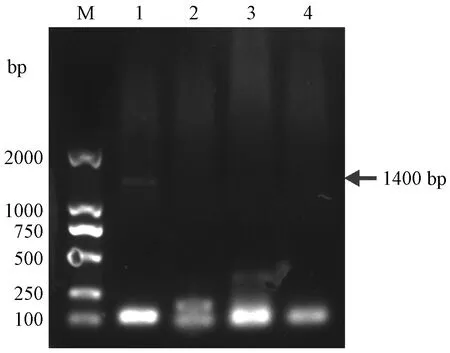
注:1,DL 2000 Marker。2~4,注射打靶载体胚胎PCR产物。图8 注射打靶载体胚胎的PCR鉴定Note. 1, DL 2000 marker. 2~4, PCR identification products of embryos injected with targeting vectors.Figure 8 PCR identification of embryos injected with targeting vectors
3 讨论
由工程化核酸酶引发的DNA双链断裂(double-strain break,DSB)被认为是产生定向突变的强有力工具,例如锌指核酸酶(ZFN)和转录激活因子样效应物核酸酶(TALEN)等[11-12],不过CRISPR/Cas9的出现,为基因的定点修饰提供了一个更加简单有效的方法[13-14]。相比于传统转基因手段使用外源基因进行原核注射的方式生产转基因动物带来的整合位点和拷贝数不确定等问题,工程化核酸酶系统可以在基因靶位点上产生精准突变。Rosa26位点最初是由基因诱捕载体Rosaβ-geo与26号转染ES细胞发生整合而被发现[15]。并被广泛用于基于ES细胞的基因打靶操作。该基因位于小鼠基因组的第6号染色体上,由3个外显子组成,可在所有细胞类型和发育阶段实现广泛表达。Rosa26的转录产物是一种非编码、非必需的核RNA,不会翻译成为蛋白质[16]。也因此,Rosa26基因位点被称为“安全港”,常被用做外源基因整合位点来实现常规和条件性的基因表达。近几年,不断有利用Rosa26位点生产转基因动物的报道出现[17-18]。本研究同样也采用了Rosa26位点作为基因打靶的位点。在CRISPR/Cas9体系中sgRNA的设计是基因编辑成功的关键,因此通常在注射之前,需要先在体外对靶DNA位点进行酶切来检验sgRNA可用性和Cas9蛋白的活性,本研究中设计构建的Rosa26的sgRNA的体外酶切实验结果表明,体外转录的sgRNA可以正常发挥编辑作用,可用于后续实验,在此基础上,我们进行了显微注射,并先后得到了Rosa26位点的基因编辑胚胎和定点突变小鼠,从而初步建立了基于CRISPR/Cas9的基因编辑体系,验证了整个体系的有效性。
组织特异性表达外源基因在研究基因功能、细胞分化和再生医学等领域具有重要作用,通过制备转基因动物产生组织特异性动物模型是常用的研究方法之一。组织特异性启动子可以带动外源基因在特定组织中表达,因此,通过Rosa26位点打靶敲入肌肉特异性表达的CRISPR/Cas9载体,就可以实现在肌肉组织中特异表达Cas9蛋白,为后续产生肌肉特异性表达Cas9的动物模型及相关研究奠定基础。常用的骨骼肌特异性启动子有α-actin启动子和合成肌肉启动子(synthetic muscle promoters SP),其中,α-actin启动子在非骨骼肌细胞中没有活性,其组成结构包含E-box和SRE调控区,可以被IGF-I和MyoG直接或间接的激活;1999年,Li等[19]根据骨骼肌α-actin的结构特性,对肌肉增强子进行分析并利用顺式作用元件和作用调控因子相对位置,人工设计了表达效率远高于天然肌源性启动子和病毒启动子的启动子序列-合成肌肉启动子SP。SP启动子由SRE、TEF-1、MEF-1和MEF-2等4种作用序列元件组成,由于这些元件的存在使得SP在已分化的肌肉组织中的转录能力得到极大增强。在研究室前期工作中,我们人工合成了SP启动子并连接到了克隆载体,在本研究中,我们将SP启动子与切除了CMV启动子的px459载体连接,随后又通过PCR和同源重组的方法将核心序列与同源臂载体Donor载体连接,并获得肌肉特异性的同源打靶载体Donor-SP-px459,通过显微注射,最终获得了小鼠肌肉特异性打靶胚胎,同时进行了胚胎移植,但暂时未成功获得打靶个体。本研究中,注射了同源打靶载体的胚胎,每6个囊胚中就有1个胚胎发生整合,整合效率达到了16.7%。这可能与同源臂大小的选择和商业化蛋白的使用有关。以往研究中,同源臂的大小一般几百到几千大小都有被使用的报道,但最近有报道称同源臂在800 bp左右时打靶效率最高[20],为此我们也采用了800 bp的同源臂来构建打靶载体;此外,其他类似研究中多采用Cas9 mRNA和sgRNA的组合进行,本研究是采用商业化的Cas9蛋白进行的。
虽然通过基因敲除可以获得MSNT基因突变个体,但由MSTN基因缺失所导致的问题和不确定性尚未被解决,尤其是涉及到基因编辑动物的生物安全性问题。因此,如何在充分利用MSTN特性来获得优良性状的同时,又能兼顾生物安全性产生理想个体,是当下研究的方向和重点。因此,接下来我们将在现有打靶胚胎的基础上,通过胚胎移植获得肌肉特异表达Cas9的小鼠,并尝试在成体水平上通过病毒携带等方式实现对小鼠MSTN基因的编辑,从而达到使小鼠肌肉量增加的目的。
猜你喜欢
——紫 苏
河南农业(2024年1期)2024-01-19 01:56:54
华人时刊(2023年1期)2023-03-14 06:43:36
汉字汉语研究(2021年2期)2021-08-30 08:58:46
中国生殖健康(2020年5期)2021-01-18 02:59:48
生殖医学杂志(2020年12期)2020-12-22 03:26:18
浙江医学(2020年17期)2020-09-21 09:06:30
中国生殖健康(2018年5期)2018-11-06 07:15:38
河北书画研究(2016年3期)2016-04-28 08:55:35
新疆医科大学学报(2015年10期)2015-12-26 12:33:30
中国医学科学院学报(2015年5期)2015-03-01 04:03:36
