
Cu-Pt-Au三元合金催化水煤气变换反应的密度泛函研究
2019-06-27 02:38:44薛继龙夏盛杰倪哲明
燃料化学学报 2019年6期
薛继龙, 方 镭, 罗 伟, 孟 跃, 陈 涛, 夏盛杰,*, 倪哲明,*
(1. 浙江工业大学 化学工程学院, 浙江 杭州 310014;2. 湖州师范学院 生命科学学院, 浙江 湖州 313000)
氢能因其清洁、高效、安全和可再生等特点,被视为二十一世纪最具发展潜力的新能源。以往的H2都由天然气经重整反应得到[1],重整气中含有的CO易导致催化剂发生中毒现象,然而,水煤气变换反应(WGSR)不仅能够减低CO浓度,还可以提高重整气中H2的含量。如何高效的催化WGSR也成为了世界各国的研究热点。且WGSR在氨合成、质子膜燃料电池(PEMFC)、甲醇合成、煤气化等方面都有广泛的应用[2-4]。现今WGSR催化剂主要的研究热点是Pt-Au双金属催化剂,因其双功能机制和电子效应能够有效地提高催化活性和耐毒性,并且能克服CO的截面尺寸效应[5]而受到了广泛的关注。如Guo等[6]研究发现,Pt-Au合金相较于Pd/C和Pt/C具有更好的稳定性和反应活性,并且对CO表现出了较强的耐毒性。Thuy-Duong等[7]比较了不同含量的Au/Pt同时负载在CeO2和TiO2时对WGSR的催化活性,发现当Au/Pt负载量都为5%时,300 ℃以下CO的转换率接近90%,表明该催化剂对WGSR具有较佳的低温催化能力。Yu等[8]制备的 Pt-Au/CeO2材料对CO转化反应具有较高的活性,在250 ℃下的转化率为78%,对WGSR的良好催化活性可能是由于负载Au和Pt之后能产生更多的活性位点。
为了提高催化剂活性及降低成本,考虑引入对WGSR也有较好催化活性的Cu[9,10]以减少催化剂中Pt和Au的含量。相比二元合金,三元合金具有更多调整自身性质的可能性。三元合金常用共电沉积方法制备[11]。例如Wang等[5]采用两步法在玻碳电极(GCE)上制备了Cu-Pt-Au三元催化剂,且发现Cu-Pt-Au三元合金材料中存在合金化微结构,当Cu∶Pt∶Au物质的量比为1.82∶1.11∶1时,催化剂对甲醇电氧化具有较优的催化活性,表明Cu-Pt-Au三元合金催化剂具有用于直接甲醇燃料电池的巨大潜力。Sohn等[12]发现,Pt1-xAuxCu3/C对氧还原反应的活性为Pt/C材料的2.4倍,且Pt0.97Au0.3Cu3表现出较优的电催化活性、质量比活性和面积比活性。Brault等[13]利用密度泛函理论(DFT)对Pt70Pd15Au15和Pt50Pd25Au25催化剂生长进行了分子动力学的模拟,在构成团簇结构时,Au层会自发包覆在表面并最终形成Pt-Pd-Au的核壳结构,发现随着Au原子含量从15%升高到25%时,催化剂的氧还原反应活性也逐渐提高。Guo等[14]利用DFT研究了PtRuM(M=Fe、Ni、Cu、Mo、Sn)三元合金团簇上O2解离的反应机理,通过反应能垒表明三金属团簇的催化性能优于双金属团簇,对O2催化解离的效果更佳。Zhang等[15]通过一步化学还原法在石墨烯片(G)上合成了具有良好分散性的AuPtPd三元合金纳米粒子,发现AuPtPd/G对甲醇氧化反应具有优异的催化活性。Hong等[16]利用非均相及外延生长方法制备了树枝状的Au/Pt和Au/PtCu纳米线,Au/PtCu较Au/Pt而言对甲醇电氧化表现出更优的电催化活性。
以上研究表明,Cu-Pt-Au三元合金催化剂可能对甲醇氧化等反应具有较优的催化活性,但其对水煤气变换反应的催化活性的研究报道极少。为探讨不同掺杂量的三元催化剂对水煤气变化反应机理的影响,本研究设计了Cux-Pty-Au(111)(x,y=1、2、3)三元合金催化剂,利用DFT理论[17]对WGSR中的小分子吸附情况及反应机理进行研究,并且阐明WGSR三条机理在三元合金催化剂上的反应历程,以期能够为WGSR在实验上设计出更加高效的催化剂提供理论依据。
1 计算方法和模型
本研究均在Dmol3模块下进行[18,19],采用广义梯度积分和Perdew-Burke-Ernzerhof泛函相结合(GGA-PBE)的密度泛函理论方法。为了减小范德华力引起的吸附误差,本研究运用了DFT-D的方法。且催化剂金属内层电子选用有效核势赝势(ECP)代替,价电子波函数采用双数值基加轨道极化函数(DNP)展开。Brillouinzone积分的k点取样采用Monkhorst-Pack的自动生成方法,网格参数设为Medium,Methfessel-Paxton smearing设置为0.005 eV。结构优化以能量差异小于2×10-5Ha、原子位移小于5.0×10-4nm以及各个原子上力的收敛4.0×10-2Ha/nm为判据。金属周期性平板结构的自旋极化现象对吸附构型及催化剂本身的性质影响很小,计算时选择自旋极化不受限制[20]。选择稳定吸附构型的反应物和产物分别作为各基元反应的初态(IS)和终态(FS),执行单个LST极大值计算并重复使用共轭梯度法优化和QST极大值进行过渡态(TS)搜索,得出其过渡态结构。精度设置为Medium,RMS convergence 精度为0.1 Ha/nm。
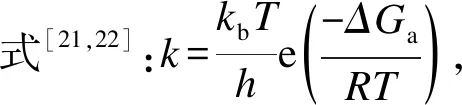
基于课题组之前在M-Au(111)二元合金表面上的研究[23],且Wu等[24]考察了不同比例的Cu-Pt同时掺杂在Au基催化剂上时的情况,发现Cu∶Pt比为1∶1时,催化剂的能量最低既结构最为稳定。当Au、Cu和Pt分别负载在CeO2等载体上时,对WGSR都有较好的催化活性[7]。考虑计算的精确度及效率,本研究构建Cu∶Pt=1∶1的3×3周期性平板模拟Cux-Pty-Au(111)(x,y=0、1、2、3)面,图1为已经优化好的Cu3-Pt3-Au(111)催化剂模型。
2 结果与讨论
2.1 催化剂的性质
2.1.1 催化剂的稳定性
多元催化剂的结构稳定性和热力学稳定性对催化活性有较为显著的影响。原子尺寸因素ΔR可以很好的对催化剂结构稳定性进行衡量[25]。当ΔR≤ 0.15时,原子之间能够形成较为稳定的合金,且ΔR越小,引起合金的晶格畸变越小[26]。而多元催化剂的结合能ΔEb可以从热力学角度衡量催化剂的稳定性,结合能越大表明多元合金催化剂的热力学稳定性越好[27]。本研究定义原子尺寸因素[28]ΔR1=|RCu-RAu|/RAu,ΔR2=|RPt-RAu|/RAu,ΔR3=|RCu-RPt|/RPt。式中,RAu、RCu、RPt分别代表Au、Cu、Pt的原子半径,分别为0.179、0.157、0.183 nm。ΔR1、ΔR2、ΔR3分别表示Cu-Au、Pt-Au、Cu-Pt二元合金的原子尺寸因素。计算得ΔR1、ΔR2、ΔR3分别为 0.123、0.022、0.142。由此可得,三种合金都可以稳定存在,且Pt-Au合金最为稳定。
本研究中定义结合能ΔEb=|Esurface-aEAu-bECu-cEPt|,其中,aEAu代表a个Au原子的能量,bECu代表b个Cu原子的能量,cEPt代表c个Pt原子的能量Esurface代表催化剂平面的总能量。结合能数据见表1。当Cu、Pt分别掺杂在Au(111)面上时,结合能都逐渐增大,当掺杂量相同时(即x=y),ΔEb[Pty-Au(111)]>ΔEb[Cux-Au(111)]。其中,ΔEb[Cu3-Au(111)]为72.97 eV,而ΔEb[Pt3-Au(111)]为77.15 eV。因此,Pt-Au合金催化剂较Cu-Au催化剂具有更好的热力学稳定性。并且Cux-Pty-Au(111)面的结合能都大于二元金属表面,其中,Cu3-Pt3-Au的结合能最大,为77.99 eV。

表1 不同掺杂比例催化剂的结合能
2.1.2 态密度图分析
d带中心可以用于衡量过渡金属催化剂表面的电子活性。d带中心距离费米能级(X= 0 eV)越近,d带空穴越多,催化剂的电子活性越高[29],进而可以通过d带中心的大小来检测和验证不同金属表面的吸附能力。态密度分析见图2,掺杂量不同的催化剂的d带中心见表2。
由表2可知,二元Pt-Au合金的d带中心明显都要比Cu-Au合金的d带中心更加靠近费米能级,具有更优的电子活性,与文献报道的一致[30]。在二元合金中,随着掺杂量的增加,合金催化剂的d带中心都逐渐向费米能级移动,其中,Cu3-Au(111)和Pt3-Au(111)面的d带中心值为-3.24、-3.18 eV。在三元合金中,当y值相同时,Cu3-Pty-Au(111)的d带中心比 Cu1-Pty-Au(111)更加靠近费米能级0.09 eV,当x值相同时,Cux-Pt3-Au(111)面较Cux-Pt1-Au(111)面的d带中心更靠费米能级0.12 eV。由此可得,Pt掺杂量的增加更有利于提高Cu-Pt-Au三元合金催化剂的电子活性,其中,Cu3-Pt3-Au(111)的d带中心为-3.05 eV,电子活性最佳。

图 2 不同金属平板的分波态密度图

表 2 不同掺杂比例Au(111)的d带中心
2.1.3 不同平板上CO和H2O的吸附
在15种催化剂表面上计算了CO和H2O的吸附能及键长。由表3可得,Cu3-Pt3-Au(111)面上CO和H2O的最优吸附构型最为稳定,并且C-O及O-H之间的键长都符合实验文献所述[31]。CO分子在Cu3-Pt3-Au(111)面上的吸附最为稳定,吸附能为2.32 eV,相较于其他Cu-Au、Pt-Au等二元催化剂来说,CO在Cux-Au(111)面上的吸附最弱,但在引入了Pt元素后,对CO的吸附能明显增大,可能是由于C中的电子向过渡金属表面转移,使C-O键被削弱,从而加强了分子在表面的吸附。综上可知,对不同催化剂表面的结合能、稳定性分析、态密度图分析可知,Cu3-Pt3-Au(111)面在所研究的催化剂中结合较为稳定,且d带中心更加靠近费米能级,具有更优越的电子活性。故在以下的讨论中选取Cu3-Pt3-Au(111)来模拟三元合金催化剂表面,计算了CO及H2O小分子在表面吸附时的电荷分布,并探讨了水煤气变换反应在Cu3-Pt3-Au(111)面上的反应机理。
2.2 反应机理
2.2.1 Cu3-Pt3-Au(111)表面的电荷布局分析
在吸附体系中,Mulliken电荷布居分析[32.33]可反映出物质中的原子与键在吸附前后的电子分布,电子转移数越多则说明小分子与金属表面的相互作用更强,吸附能更大。为了分析CO及H2O在Cu3-Pt3-Au(111)面上的吸附情况,对CO及H2O分子在表面的最优吸附构型进行了Mulliken电荷布居分析,具体见表4。

表4 Mulliken电荷布局分析
吸附之前CO小分子显电中性,其中,O原子的电负性大于C原子,因此,在吸附前的CO分子中,成键电子尤其是p电子倾向于O原子,所以O原子带负电荷,C原子带正电荷。CO的整体电荷从0.000 e分别增加为0.433 e,H2O的整体电荷从0.000 e增加为0.123 e,说明在吸附过程中,部分电子从CO、H2O分子转移到了金属表面,增强了CO及H2O在催化剂表面的吸附。
2.2.2 WGSR中小分子在Cu3-Pt3-Au(111)表面上的吸附
为了明确WGSR中各个小分子在催化剂表面上的吸附程度,本研究引入吸附能(Eads)来衡量Eads=E(A/surface)-EA-Esurface,其中,E(A/surface)为小分子吸附在催化剂表面上时体系的总能量,EA表示小分子的能量,Esurface表示Cu3-Pt3-Au(111)面的总能量。Eads的符号和数值大小可衡量发生吸附的可能性和吸附作用的强弱,其值越大表示吸附过程中放出热量越多,形成的吸附体系越稳定。水煤气变换反应中共有10个吸附物质,分别为:CO、H2O、OH、H、O、COOH、CO2、H2、HCO、O和CHO,在 Cu3-Pt3-Au(111)面吸附位的单点吸附共有70种。分别将10个吸附物质在Cu3-Pt3-Au(111)面上吸附的结构进行优化计算,通过对Eads的比较,得到10个吸附物种在催化剂上的最优吸附构型。吸附能如表5所示,10种吸附小分子在Cu3-Pt3-Au(111)面的Cu-Pt-Bri和Fcc处的吸附能都普遍较大。当CO以碳端吸附在Fcc的时候吸附能最大值为2.32 eV,H2O吸附在Cu-Pt-Bri时的吸附能最大,其值为0.54 eV,并且CO和H2O分子的键长都稍有拉长,其中,碳氧键拉长了0.0047 nm,O-H键拉长了0.0009 nm。并且在Pt原子附近的吸附位置都明显较Cu原子附近的吸附位的吸附能更大,所得到的吸附构型更加稳定。

表5 Cu3-Pt3-Au(111)表面上小分子的吸附能
2.2.3 WGSR机理
水煤气变换反应可按氧化还原机理、羧基机理、甲酸机理三种机理进行[34],并计算了WGSR中各个基元反应的能垒及反应热,见表6,具体反应路径见图3。
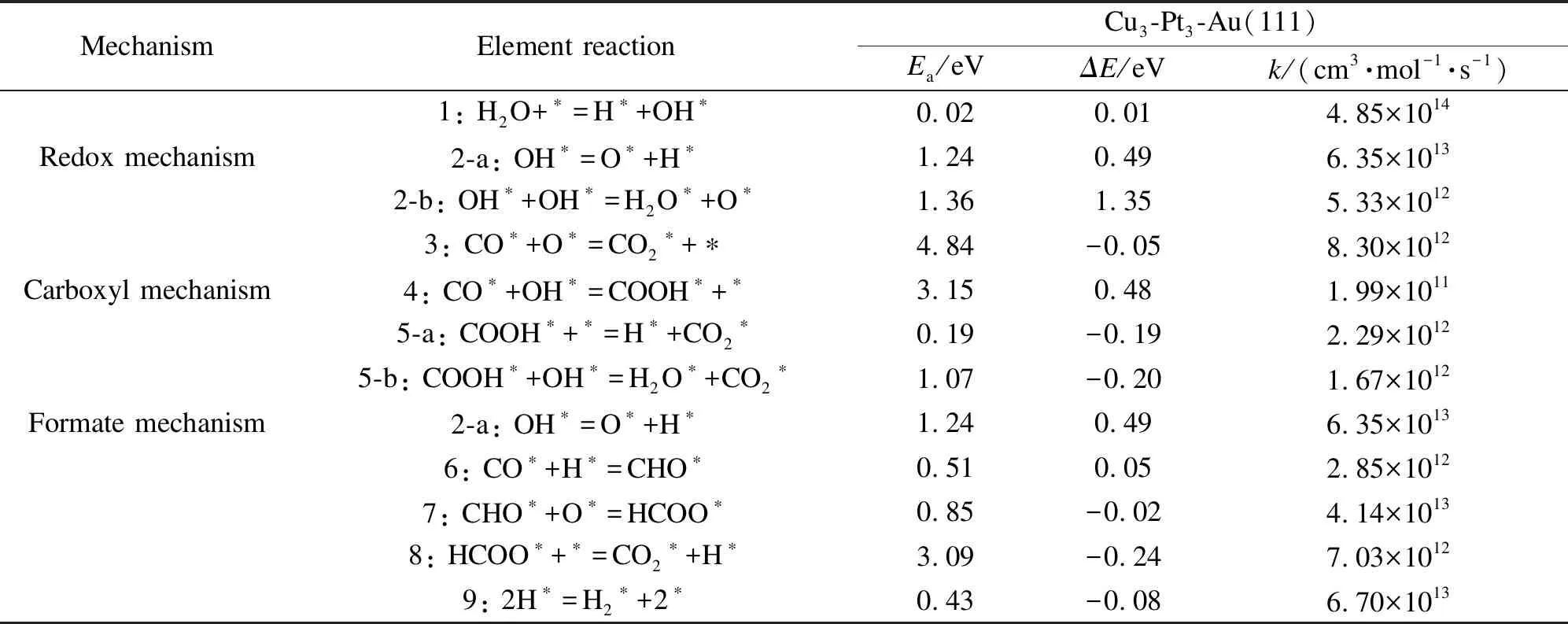
表6 Cu3-Pt3-Au(111)表面上各基元反应的活化能(Ea)、反应热(ΔE)和速率常数k
由图3可知,在氧化还原机理中,水解离后产生的OH经过2-a继续解离。以O端竖直吸附在Cu-Pt-Bri位上的OH解离成为游离的O、H,并平行吸附在Cu-Pt-Bri位的两侧,相距1.386 nm,该步骤需跨越1.24 eV的能垒,吸收热量0.49 eV,这是O原子的来源之一。另外,OH可以在催化剂表面发生歧化反应产生O,具体反应为2-b,该步骤能垒为1.36 eV,需要吸收热量1.35 eV,最终产生的H2O吸附在Top-Cu位,O原子吸附在Cu-Pt-Bri位。之后O原子在逐渐靠向CO分子,形成CO2最终吸附在Cu-Pt-Bri位上,该步骤为氧化还原机理中的速控步骤,需要跨过4.84 eV大小的能垒,并放出0.05 eV的热量,产生的CO2随后脱附,两个活性吸附位上H靠近形成H2,最终得到H2。在羧基机理中,水解离产生的OH吸附在Cu-Bri位,随后OH逐渐向Pt-Bri位置的CO靠近,形成COOH中间体,最终以C端吸附在催化剂表面,该步骤需要跨过3.15 eV能垒,且为吸热反应,吸热0.48 eV。形成CO2存在两种可能的反应,在图3的5-a中,中间体COOH继续自解离成CO2和H,CO2逐渐远离催化剂表面,其中,C原子距表面0.36145 nm,该反应需克服0.19 eV的能垒,并放出0.19 eV的热量。但在图3的5-b中,COOH中的O-H断裂,H向吸附在Cu-Bri位置上的OH逐渐靠近,形成的H2O脱附,OCO中间体则转化为CO2,并且图3的5-b中的反应产物都逐渐远离催化剂表面,最终脱附。
在甲酸机理中,CO与一个H原子相互靠近形成CHO中间产物并以C、O平行吸附在Fcc位,该基元反应需克服0.51 eV的能垒,并吸热0.05 eV。之后O向CHO中的C端移动,最终形成C-O键,构成中间产物HCOO,能垒为0.85 eV,该反应为放热反应,释放0.02 eV热量。在反应8中HCOO发生自解离,解离出的CO2在表面脱附,H则和其他活性位点上的H结合成为H2,进而脱附,该步需克服3.09 eV的能垒,并放热0.24 eV。具体的过渡态见图4所示。各机理能量变化如图5所示。

图 3 WGSR机理示意图

图 4 甲酸机理中的过渡态

图 5 各机理在Cu3-Pt3-Au(111)上的能量变化
3 结 论
本研究计算了不同掺杂量的二元合金(Cu-Au,Pt-Au)及三元合金(Cu-Pt-Au)的性质,表明在二元合金中Pty-Au较Cux-Au具有更好的稳定性,当x=y时,ΔEb[Pty-Au(111)]>ΔEb[Cux-Au(111)]成立。Pty-Au(111)与Cux-Au(111)相比较而言其d带中心更靠近费米能级,表现更优的电子活性。当再引入第三金属构成Cu-Pt-Au催化剂时,稳定性有较大提高,Cu3-Pt3-Au(111)面最为稳定,结合能为77.99 eV,结合能大于二元合金催化剂,且Cu3-Pt3-Au(111)的d带中心最靠近费米能级,为-3.05 eV,电子活性最佳。
分析了15种不同金属平面对CO及H2O的吸附构型及吸附能,发现Cu3-Pt3-Au(111)对CO及H2O有较强的吸附,更有利于水煤气变换反应进行。
讨论了WGSR中三条机理在Cu3-Pt3-Au(111)上的反应历程,发现在三元合金表面水解离反应极易发生。而反应3、反应4和反应8则分别作为氧化还原机理、羧基机理和甲酸机理中的速控步骤,其中,氧化还原机理中形成TS3需要克服的能垒值最大,为4.84 eV,而羧基机理和甲酸机理的能垒分别为3.15、3.09 eV。甲酸机理相对比其他两种机理而言更容易在Cu3-Pt3-Au(111)上发生反应,具体反应路径为:1→2-a→6→7→8→9。并发现存在CO或OCO中间体的基元反应能垒值较大,可能是Pt与CO的强吸附作用。
猜你喜欢
燃料化学学报(2023年3期)2023-03-11 03:34:40
大学物理(2022年9期)2022-09-28 01:10:52
中学课程辅导·教学研究(2021年8期)2021-07-14 13:44:52
燃料化学学报(2021年5期)2021-06-02 14:01:38
物理通报(2020年7期)2020-07-01 09:28:02
原子与分子物理学报(2015年3期)2015-11-24 12:49:34
文理导航(2015年26期)2015-09-29 14:12:24
淮南师范学院学报(2015年3期)2015-03-22 01:16:19
河北科技大学学报(2015年5期)2015-03-11 16:16:34
无机化学学报(2014年4期)2014-02-28 17:31:23
