
HPLC-MS/MS法手性拆分泮托拉唑钠对映体及大鼠血浆中药代动力学研究
2019-06-10 07:16:34袁艳娟乔红群
质谱学报 2019年3期
袁艳娟,刘 晶,邵 卿,乔红群
(南京工业大学,江苏省药物研究所,江苏省药物安全性评价中心,江苏 南京 211800)
泮托拉唑是一种选择性质子泵抑制剂(PPIs),治疗酸相关的胃肠道疾病。所有PPIs,包括奥美拉唑、泮托拉唑、兰索拉唑等,都具有手性苯并咪唑亚砜结构,现临床上使用药物多为其消旋体结构[1]。手性化合物的拆分通常在正相色谱条件下可以获得较好的分离效果,但是,反相手性色谱具有毒性小、成本低的优点,并与实际样品具有很好的相容性。现已报道的泮托拉唑钠手性分离方法包括高效液相色谱-紫外检测(HPLC-UV)法[2-5]和多维色谱分离法[6],但是这些方法的选择性差、灵敏度低、耗时相对较长。因此,有必要开发一种全新的量化方法对血浆中泮托拉唑钠对映体进行手性拆分。高效液相色谱-串联质谱法(HPLC-MS/MS)越来越多地应用于手性药物分离。Chen等[1]用APCI源手性分离泮托拉唑对映体;程艳玲等[7]、曹晓丽等[8]通过HPLC-MS/MS法,ESI+模式,使用手性色谱柱检测犬血浆中泮托拉唑对映体。
本研究拟建立Chiralpak IE色谱柱手性拆分泮托拉唑对映体的HPLC-MS/MS方法,采用ESI-模式分离大鼠血浆样本中左右旋泮托拉唑。同时,按照《中国药典》2015年版附录“生物样本定量分析方法验证指导原则”进行方法学验证[9],并应用该方法研究大鼠单剂量药代动力学。
1 实验部分
1.1 仪器与试剂
Agilent 1260高效液相色谱仪:美国Agilent公司产品,配有在线真空脱气机、自动进样器、柱温箱;API4000+三重四极杆质谱仪、电喷雾离子化源:美国AB Sciex公司产品,配有Analyst1.6.2软件;Chiralpak IE色谱柱:大赛璐药物手性技术上海有限公司产品。
左旋泮托拉唑钠(纯度99.9%)、右旋泮托拉唑钠(纯度99.8%):陕西合成药业有限公司产品;双氯芬酸钠:由中国食品药品检定研究院提供; SD大鼠:由北京维通利华实验动物技术有限公司提供;乙腈、甲醇、甲酸:均为美国Tedia公司产品;实验用水为Millipore超纯水。
1.2 实验条件
1.2.1色谱条件 色谱柱:Chiralpak IE柱(4.6 mm×250 mm×5 μm);流动相:A为0.1%甲酸水溶液,B为乙腈;等度洗脱:A-B(30∶70,V/V);流速:0.9 mL/min;柱温:室温;进样量:5 μL。
1.2.2质谱条件 电喷雾离子源负离子模式(ESI-);电喷雾电压(IS)-4 500 V;温度550 ℃;源内辅助气1(GS1,N2)压力344.7 kPa,源内辅助气2(GS2,N2)压力344.7 kPa;气帘气(CUR,N2)压力172.4 kPa;碰撞气(CAD,N2)压力48.3 kPa;解簇电压(DP):左、右旋泮托拉唑-60 V,内标双氯芬酸钠-45 V;碰撞能量(CE):左、右旋泮托拉唑-16 V,内标双氯芬酸钠-14 V;扫描方式为多反应监测模式(MRM);用于定量分析的离子对:左、右旋泮托拉唑m/z382.0>229.8,内标双氯芬酸钠m/z293.9>249.6。
1.3 标准溶液的配制
精密称取适量的左、右旋泮托拉唑钠对照品,分别置于10 mL容量瓶中,甲醇溶解并定容,配制成1 g/L的左旋泮托拉唑钠、右旋泮托拉唑钠标准储备液。
称取适量的双氯芬酸钠对照品,置于10 mL容量瓶中,用乙腈-水溶液(50∶50,V/V)定容,得1 g/L内标储备液。用乙腈稀释成浓度为500 μg/L的溶液,作为内标工作液。
1.4 动物实验
选用SD大鼠,实验前禁食12 h,不禁水,静脉推注给药,剂量为低(4 mg/kg)、中(8 mg/kg)和高(16 mg/kg)。同时设立右旋对照组(8 mg/kg)和消旋对照组(16 mg/kg),每组6只动物,雌雄各半。通过大鼠眼底静脉丛采集全血(每只动物约0.2~0.4 mL/次),采血时间为给药前0 min,给药后第2、5、15、30、60、90、120、150、180、240、300 min。血液采集至肝素钠(10 U)抗凝的EP管中,充分摇匀,离心后取出血浆,于-20 ℃保存,待测。
1.5 大鼠血浆样本预处理
待测血浆样品(-20 ℃)于室温下自然解冻后,精密吸取25 μL,置于1.5 mL离心管中,精密加入150 μL内标溶液(500 μg/L双氯芬酸钠),涡旋3 min,以16 000 r/min离心5 min,取150 μL上层溶液至1.5 mL离心管中,再次以16 000 r/min离心5 min,取5 μL上层溶液进样分析。
2 结果与讨论
2.1 HPLC-MS/MS条件优化
本研究对检测模式及色谱条件进行优化,分别考察了ESI+、ESI-模式下,大鼠血浆样本中泮托拉唑钠对映体的手性拆分。结果表明,ESI+模式下基质效应比较明显;而ESI-模式无基质效应,灵敏度高,定量限可达5 μg/L,能够满足本研究检测要求。
以5 mmol/L甲酸铵水溶液、5 mmol/L乙酸铵水溶液、0.1%甲酸水溶液、0.1%乙酸水溶液与甲醇、乙腈组成流动相体系,考察左、右旋泮托拉唑的分离效果。结果表明,左、右旋泮托拉唑在乙腈体系下的峰形及响应明显优于甲醇体系。5 mmol/L甲酸铵水溶液、5 mmol/L乙酸铵水溶液-乙腈体系在梯度条件下和0.1%甲酸水溶液-乙腈体系在等度条件下均能较好地分离血浆中左、右旋泮托拉唑。但因等度洗脱条件较梯度条件简单,分析时间短,本实验最终选择0.1%甲酸水溶液-乙腈等度洗脱体系。
2.2 血浆样本预处理条件优化
本实验考察了液液萃取和蛋白沉淀方法对血浆样本进行预处理。分别采用乙酸乙酯、乙醚、正己烷、甲基叔丁基醚作为萃取剂,乙腈、甲醇作为沉淀剂,进行血浆样本预处理优化。结果表明,正己烷不能将泮托拉唑从血浆中提取出来,乙醚和甲基叔丁基醚的提取回收率约为80%,而使用乙酸乙酯作为萃取剂时,提取回收率大于85%。甲醇作为沉淀剂时存在基质干扰,乙腈则无基质干扰,提取回收率与乙酸乙酯液液萃取效率相当。因此,本实验采用乙腈蛋白沉淀方法进行血浆样本预处理。
2.3 选择性研究
取6只大鼠的空白血浆,按2.2节方法处理,在1.2.1节条件下分别检测空白血浆(图1)、空白血浆添加左旋和右旋泮托拉唑钠(图2~3)、空白血浆添加内标(图4)的特异性。由图1~4可见,左旋、右旋泮托拉唑保留时间分别为6.43 min和5.03 min,二者均能实现基线分离,内标保留时间为4.63 min,各检测物峰形良好。血浆中生物基质对左旋和右旋泮托拉唑无干扰,方法具有较高的特异性。
2.4 标准曲线及定量下限
使用空白血浆配制左旋、右旋泮托拉唑浓度分别为5、10、20、50、100、200、500、1 000、2 000和5 000 μg/L的血浆样本,按2.2节方法操作,进行色谱分析。经验证,在该实验条件下,左旋和右旋泮托拉唑血浆样品的线性范围均为5.0~5 000.0 μg/L,最低定量限均为5.0 μg/L。

注:a.左、右旋泮托拉唑离子对;b.内标离子对图1 空白血浆图谱Fig.1 Chromatograms of blank plasma
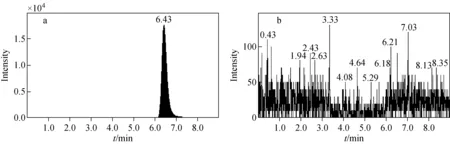
注:a.左、右旋泮托拉唑离子对;b.内标离子对图2 血浆样本中添加左旋泮托拉唑图谱Fig.2 Chromatograms of plasma samples supplemented with L-pantoprazole sodium
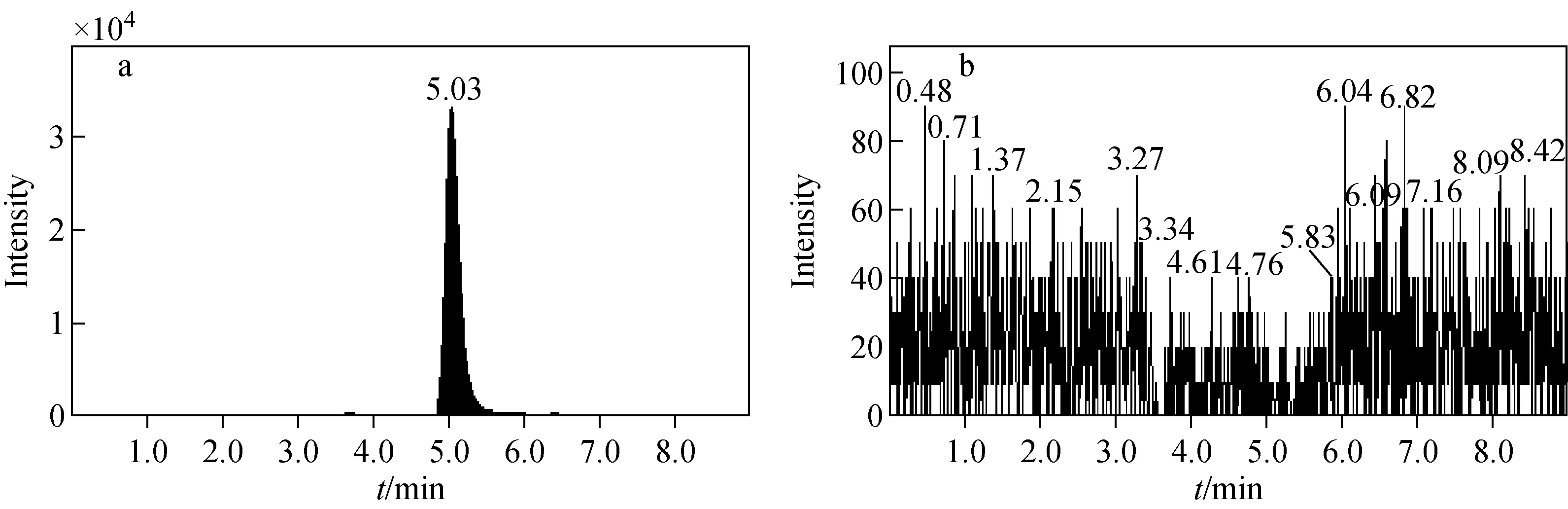
注:a.左、右旋泮托拉唑离子对;b.内标离子对图3 血浆样本中添加右旋泮托拉唑图谱Fig.3 Chromatograms of plasma samples supplemented with D-pantoprazole sodium

注:a.左、右旋泮托拉唑离子对;b.内标离子对图4 空白血浆样本中添加内标图谱Fig.4 Chromatograms of blank samples with internal standard
2.5 基质效应实验
取空白溶液及分别来源于6只大鼠的空白血浆,配制成左旋、右旋泮托拉唑浓度分别为10、50、500、2 000 μg/L的无基质及基质样本,考察基质效应。检测得到峰面积分别为A2、A1,详细结果列于表1。左旋泮托拉唑基质效应分别为96.4%、96.8%、107.2%、109.4%,右旋泮托拉唑基质效应分别为96.1%、98.8%、104.7%、101.7%。
2.6 提取回收率实验
取大鼠空白血浆,配制成左旋、右旋泮托拉唑浓度分别为10、50、500、2 000 μg/L的血浆样本,考察提取前后的回收率,结果列于表2。左旋泮托拉唑的方法回收率为96.4%、106.8%、97.9%、114.3%,右旋泮托拉唑的方法回收率为112.8%、105.2%、91.1%、108.6%。

表1 左旋泮托拉唑、右旋泮托拉唑、内标在大鼠血浆中的基质效应(n=6)Table 1 Matrix effects of D, L-pantoprazole and internal standard in rat plasma (n=6)
注:A1、A2分别为右旋、左旋泮托拉唑的峰面积

表2 左旋泮托拉唑、右旋泮托拉唑、内标在大鼠血浆中的提取回收率(n=6)Table 2 Recoveries of D, L-pantoprazole and internal standard in rat plasma (n=6)
2.7 精密度和准确度实验
按2.4节方法配制10、50、500、2 000 μg/L四种不同浓度的血浆样本各5份,按2.2节方法操作,一天配制一批精密度样品及一条随行标准曲线,连续测试3天,共3批,结果列于表3~4。结果表明,批间和批内精密度均小于10%,批间和批内准确度在85%~115%之间。
2.8 稳定性实验
按照2.4节方法配制左旋和右旋泮托拉唑的低(10 μg/L)、中(50 μg/L)、高(2 000 μg/L)三个浓度血浆样本,反复冻融3次,于37 ℃水浴解冻后,提取测定药物浓度,考察反复冻融稳定性。将血浆样本放置于室温下24 h,提取测定其药物浓度,考察血浆样本放置稳定性。将提取的样品于4 ℃放置48 h,测定其浓度,考察进样瓶中放置稳定性。将含药血浆置于-20 ℃冰箱中冷藏9个星期,提取测定药物浓度,考察其长期稳定性。结果表明,血浆中左旋和右旋泮托拉唑在以上各种稳定性研究实验条件下均能保持稳定。

表3 大鼠血浆样品中左旋泮托拉唑的批内、批间精密度和准确度(n=6)Table 3 Intra- and inter-assay precision and accuracy of L-pantoprazole in rat plasma samples (n=6)

表4 大鼠血浆样品中右旋泮托拉唑的批内、批间精密度和准确度(n=6)Table 4 Intra- and inter-assay precision and accuracy of D-pantoprazole in rat plasma samples (n=6)
2.9 稀释可靠性与残留考察
配制超出线性范围浓度的含药血浆,使用空白血浆稀释2倍和100倍作为稀释样本,提取并测定其中的药物浓度。结果表明,血浆样本稀释过程对测定结果并无影响。
在标准曲线最高浓度后进行双空白样本检测,考察其残留。结果表明,血浆样本检测无残留。
2.10 大鼠药代动力学结果
大鼠静脉注射给予4、8、16 mg/kg的注射用左旋泮托拉唑钠后,平均血药浓度-时间曲线示于图5,药代动力学参数列于表5。大鼠静脉注射给予消旋对照品后,左、右旋泮托拉唑平均血药浓度-时间曲线示于图6,右旋泮托拉唑药代动力学参数列于表6。
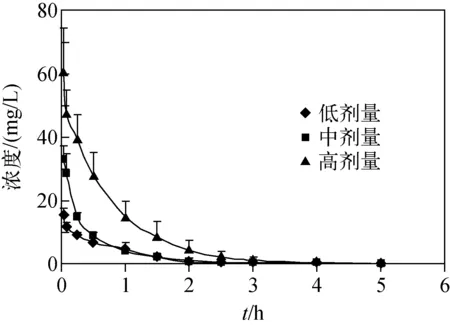
图5 左旋泮托拉唑平均血药浓度-时间曲线Fig.5 Average plasma concentration-time curve

表5 左旋泮托拉唑药代动力学参数均值(n=6)Table 5 Average of L-pantoprazole pharmacokinetic parameters (n=6)
注:t-test与低剂量组相比,*表示p<0.05,**表示p<0.01;与中剂量组相比,#表示p<0.05

图6 左、右旋泮托拉唑平均血药浓度-时间曲线Fig.6 Average plasma concentration-time curve
低、中、高剂量组药代动力学参数统计结果表明,在4~16 mg/kg剂量范围内,左旋泮托拉唑在大鼠体内的暴露随剂量增大而增加,而MRT、t1/2、Vz等参数不会随剂量出现明显的改变。
统计药代动力学参数发现,大鼠静脉给予注射用泮托拉唑钠后,左、右旋泮托拉唑在动物体内的药代动力学行为略有不同:左旋对映体暴露水平略高于右旋;右旋对映体存在时可增加左旋对映体在大鼠体内的暴露水平,降低其清除率;而左旋对映体对右旋对映体的药代动力学行为无明显影响。
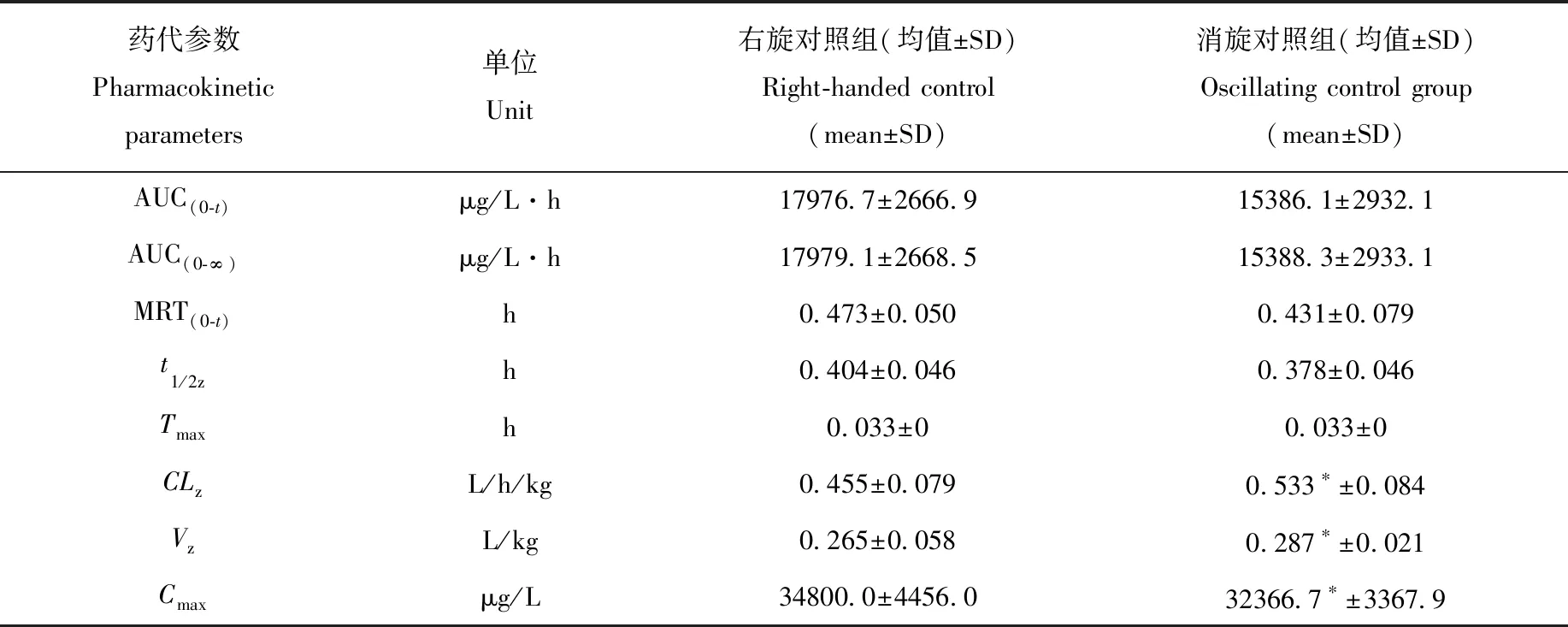
表6 右旋泮托拉唑药代动力学参数均值(n=6)Table 6 Average of D-pantoprazole pharmacokinetic parameters (n=6)
注:t-test与消旋对照组左/右对映体相比,*表示p<0.05
3 结论
本实验建立了Chiralpak IE色谱柱手性拆分泮托拉唑对映体的HPLC-MS/MS分析方法。在优化的实验条件下,左旋和右旋泮托拉唑在血浆基质下能基线分离,空白血浆无干扰,方法选择性良好,二者线性范围均为5.0~5 000.0 μg/L,最低定量限均为5.0 μg/L。经方法学验证,该方法回收率、基质效应、精密度、准确度、残留及稀释过程均符合生物样本分析要求。将该方法应用于大鼠血浆样本中左旋、右旋泮托拉唑钠药代动力学研究,结果可靠,可用于生物样本中泮托拉唑钠对映体的痕量分析。
猜你喜欢
高等学校化学学报(2024年2期)2024-03-06 06:31:12
家庭医学(下半月)(2020年3期)2020-05-30 12:42:12
家庭医学(下半月)(2019年8期)2019-09-25 09:02:08
布达拉(2019年3期)2019-06-11 05:34:00
课程教育研究(2018年1期)2018-03-31 09:28:22
中成药(2017年9期)2017-12-19 13:34:31
药学与临床研究(2015年4期)2015-06-05 11:35:53
温州医科大学学报(2014年7期)2014-07-18 02:43:24
中国药业(2014年21期)2014-05-26 08:56:51
郑州大学学报(理学版)(2013年2期)2013-03-11 20:30:30
