
蒙药乌兰三味汤散质量标准的研究
2019-06-01 02:51陈建平李鑫雨巴俊杰
中成药 2019年4期
王 敏, 布 仁, 陈建平, 李鑫雨, 巴俊杰
(内蒙古医科大学药学院, 内蒙古 呼和浩特 010110)
乌兰三味汤散, 蒙文名又称玛日布-3 汤散、 乌兰-3等, 出自《蒙医成方选》, 目前收载于《卫生部药品标准》蒙药分册[1]。 该方由紫草茸、 茜草、 枇杷叶三味药材组成,剂型为汤散剂, 有清血热之功效, 用于治疗肺热咳嗽, 肺、肾损伤性热, 痰中带血, 尿频尿痛, 膀胱刺痛, 以紫草茸为主, 可清血热及肺、 肾热, 茜草、 枇杷叶为伍, 辅以清血热, 全方性凉, 为清肺肾热之良方[2]。 周桂坤[3]和李丽[4]分别采用HPLC 法测定了其中大叶茜草素和茜草素的含有量, 嘎鲁等[5]测定了微量元素, 原标准有制法、 处方、性状、 功效, 规格、 用法、 用量及贮藏条件[1]。 本研究对该蒙药进行了显微和薄层色谱鉴别, 并用HPLC 法测定了熊果酸的含有量, 这对完善和提高其质量标准和质控水平有着重要意义。
1 仪器与材料
1.1 仪器 LC-8A 高效液相色谱仪 (日本岛津公司);AB135-S 型电子天平(瑞士梅特勒-托利多公司); 奥林巴斯pd25 摄像采集器; KQ-500DE 型数控超声波清洗器(金坛市江南仪器厂); bx41 数码显微镜; SLD-Ⅰ型薄层色谱摄像仪(天津思利达色谱技术开发公司); SHZ-CA 型循环多用真空泵(昆山市超声仪器有限公司); HH-2 恒温水浴锅(巩义市英峪仪器厂); ZDZ-6 型紫外分析仪(上海信谊仪器厂); 薄层展开槽(上海安亭电子仪器厂)。
1.2 材料 乌兰三味汤散(内蒙古蒙药股份有限公司、 内蒙古国际蒙医医院国家蒙药制剂中心和内蒙古医科大学内蒙古自治区中蒙药重点实验室)。 紫草茸、 茜草、 枇杷叶经内蒙古医科大学药用植物教研室渠弼副教授鉴定为正品,均由内蒙古国际蒙医医院国家蒙药制剂中心提供。 紫草茸、茜草、 枇杷叶对照药材(中国食品药品检定研究院, 批号分别 为121052-201302、 121049-201404、 201261-201303)。大叶茜草素、 熊果酸对照品(中国食品药品检定研究院,批号分别为110884-2-1405、 110742-201421)。 甲醇和乙腈为色谱纯; 其他试剂为分析纯; 水为超纯水。
2 方法与结果
2.1 显微鉴别 茜草为根类药材, 枇杷叶为叶类药材, 紫草茸为树胶类药材, 显微特征差别较大, 均具有不受干扰的专属性特征, 并易于察见。 将混合粉末制片后(水装片、 水合氯醛透化片、 染色片等), 置于显微镜下观察。
2.1.1 紫草茸 将本品粉末制片后在显微镜下观察, 可见黄棕色或红棕色不规则半透明块状或颗粒状物, 为紫草茸的显微特征[6]。
2.1.2 茜草 将本品粉末制片后在显微镜下观察, 可见导管为具缘纹孔导管, 其薄壁细胞含红棕色颗粒, 草酸钙针晶存在于薄壁细胞中或散在, 为茜草的显微特征[7]。
2.1.3 枇杷叶 将本品粉末制片后在显微镜下观察, 可见纤维周围薄壁细胞含方晶, 且形成晶纤维, 形状为非腺毛弯曲或呈人字形, 草酸钙簇晶直径为40 ~110 μm, 为枇杷叶的显微特征。
2.2 薄层色谱鉴别
2.2.1 紫草茸 参照文献[8] 和2015 年版《中国药典》四部通则0502 薄层色谱法[9], 称取本品1.5 g、 紫草茸对照药材0.5 g、 紫草茸阴性本品1.0 g, 制备供试品溶液、对照药材溶液、 阴性对照溶液, 点样量都为6 μL, 薄层板为硅胶G 板, 展开剂为氯仿-甲苯-丙酮-甲酸(5.0 ∶5.0 ∶0.8 ∶0.2)。 经点样、 饱和、 展开和热风吹干后, 在薄层色谱上于365 nm 紫外灯下检视, 供试品在与对照药材相对应的位置显示多个相同颜色的荧光斑点, 而阴性对照品色谱
上无干扰斑点, 见图1。
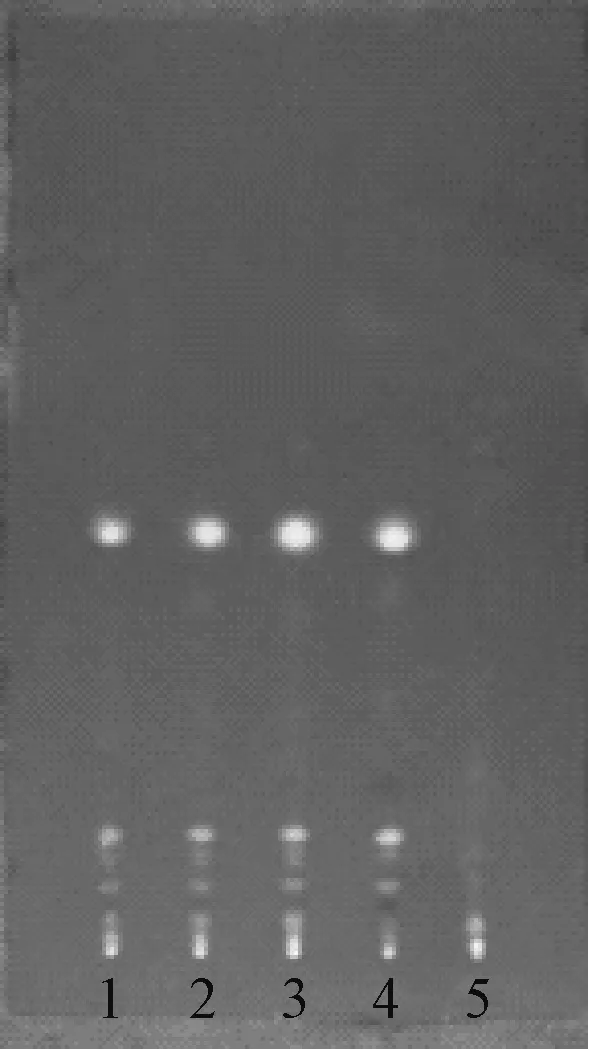
图1 紫草茸薄层鉴别
2.2.2 茜草 参照2015 年版《中国药典》 一部茜草项下薄层色谱鉴别[10]、 文献[11] 和2015 年版《中国药典》四部通则0502 薄层色谱法[9], 称取本品1.5 g、 茜草对照药材0.5 g、 茜草阴性本品1.0 g, 制备供试品溶液、 对照药材溶液、 阴性对照溶液。 另取大叶茜草素对照品, 用甲醇溶解制成每1 mL 含2.5 mg 该成分的溶液, 作为对照品溶液, 点样量都为5 μL, 薄层板为硅胶G 板, 展开剂为石油醚(60~90 ℃) -丙酮(4 ∶1)。 经点样、 饱和、 展开和热风吹干后, 于365 nm 紫外灯下检视, 供试品在与对照药材相对应的位置显示多个相同颜色的荧光斑点, 在与对照品相对应的位置显示相同的蓝色荧光斑点, 而阴性对照品色谱上无干扰斑点, 见图2。

图2 茜草薄层鉴别
2.2.3 枇杷叶 参照2015 年版《中国药典》 一部枇杷叶项下薄层色谱鉴别[12]、 文献[13] 和2015 年版《中国药典》 四部通则0502 薄层色谱法[9], 取本品3 g、 枇杷叶对照药材1 g、 枇杷叶阴性本品2 g, 制备供试品溶液、 对照药材溶液、 阴性对照溶液。 另取熊果酸对照品, 用甲醇溶解制成每1 mL 含1 mg 该成分, 作为对照品溶液, 点样量都为3 μL, 薄层板为硅胶G 板, 展开剂为甲苯-丙酮(5 ∶1), 显色剂为10%硫酸乙醇溶液。 经点样、 饱和、 展开、 显色和热风吹干后, 供试品在与对照药材相对应的位置显示2 个相同颜色的斑点, 在与对照品相对应的位置显示相同颜色的斑点, 而阴性对照品色谱无干扰斑点, 见图3。
2.3 HPLC 法测定熊果酸含有量
2.3.1 对照品溶液制备 精密称取熊果酸对照品适量, 加甲醇溶解制成0.70 mg/mL 溶液, 即得。
2.3.2 供试品溶液制备 精密称取乌兰三味汤散3.0 g,置于25 mL 量瓶中, 精密加入氯仿20 mL, 称定质量, 超声20 min, 间歇10 min, 继续超声20 min, 冷却至室温后再称定质量, 减少的质量用氯仿补足, 摇匀, 过滤, 取滤液5 mL, 在水浴锅上蒸干, 甲醇复溶, 定容至5 mL 量瓶中, 即得(进样时用0.45 μm 微孔滤膜过滤)。
2.3.3 色谱条件 ODS-2 Hypersil 柱(200 mm×4.6 mm,5 μm); 流动相乙腈-甲醇-水-乙酸铵(60 ∶14 ∶30 ∶0.6);体积流量1 mL/min; 柱温25 ℃; 检测波长215 nm。 该色谱条件下, 熊果酸色谱峰与其他组分色谱峰分离度良好,见图4。
2.3.4 线性关系考察 精密吸取“2.3.1” 项下熊果酸对照品溶液0.2、 0.4、 0.6、 0.8、 1.0 mL, 分别置于1 mL 量瓶中, 甲醇定容, 摇匀, 得到质量浓度分别为0.140 8、0.281 6、 0.422 4、 0.563 2、 0.704 0 mg/mL 溶 液, 在“2.3.3” 项色谱条件下分别进样20 μL 测定其峰面积。 以色谱峰面积为纵坐标(Y), 质量浓度为横坐标(X) 进行回归, 得回归方程为Y=5×106X+209 839 (r=0.999 3),在0.140 8~0.704 0 mg/mL 范围内线性关系良好。
2.3.5 精密度试验 精密吸取“2.3.1” 项下对照品溶液20 μL, 在“2.3.3” 项色谱条件下测定, 重复5 次, 测得峰面积RSD 为0.71%, 表明仪器精密度良好。
2.3.6 重复性试验 取本品(内蒙古医科大学内蒙古自治区中蒙药重点实验室) 5 份, 按“2.3.2” 项下方法制备供试品溶液, 在“2.3.3” 项色谱条件下测定, 测得熊果酸含有量RSD 为2.9%, 表明该方法重复性良好。
2.3.7 稳定性试验 取“2.3.2” 项下供试品溶液, 在“2.3.3” 项色谱条件下于0、 2、 4、 6、 8 h 测定, 测得峰面积RSD 为1.56%, 表明供试品溶液在8 h 内稳定性良好。
2.3.8 加样回收率试验 取本品(内蒙古医科大学内蒙古自治区中蒙药重点实验室) 6 份, 每份约1.5 g, 精密称定, 1、 2 号样品加入原含有量50%熊果酸对照品, 3、 4 号样品加入原含有量100%熊果酸对照品, 5、 6 号样品加入原含有量150%熊果酸对照品, 按“2.3.2” 项下方法制备供试品溶液, 在“2.3.3” 项色谱条件下测定。 结果, 平均加样回收率为100.1%, RSD 为1.7%, 见表1。
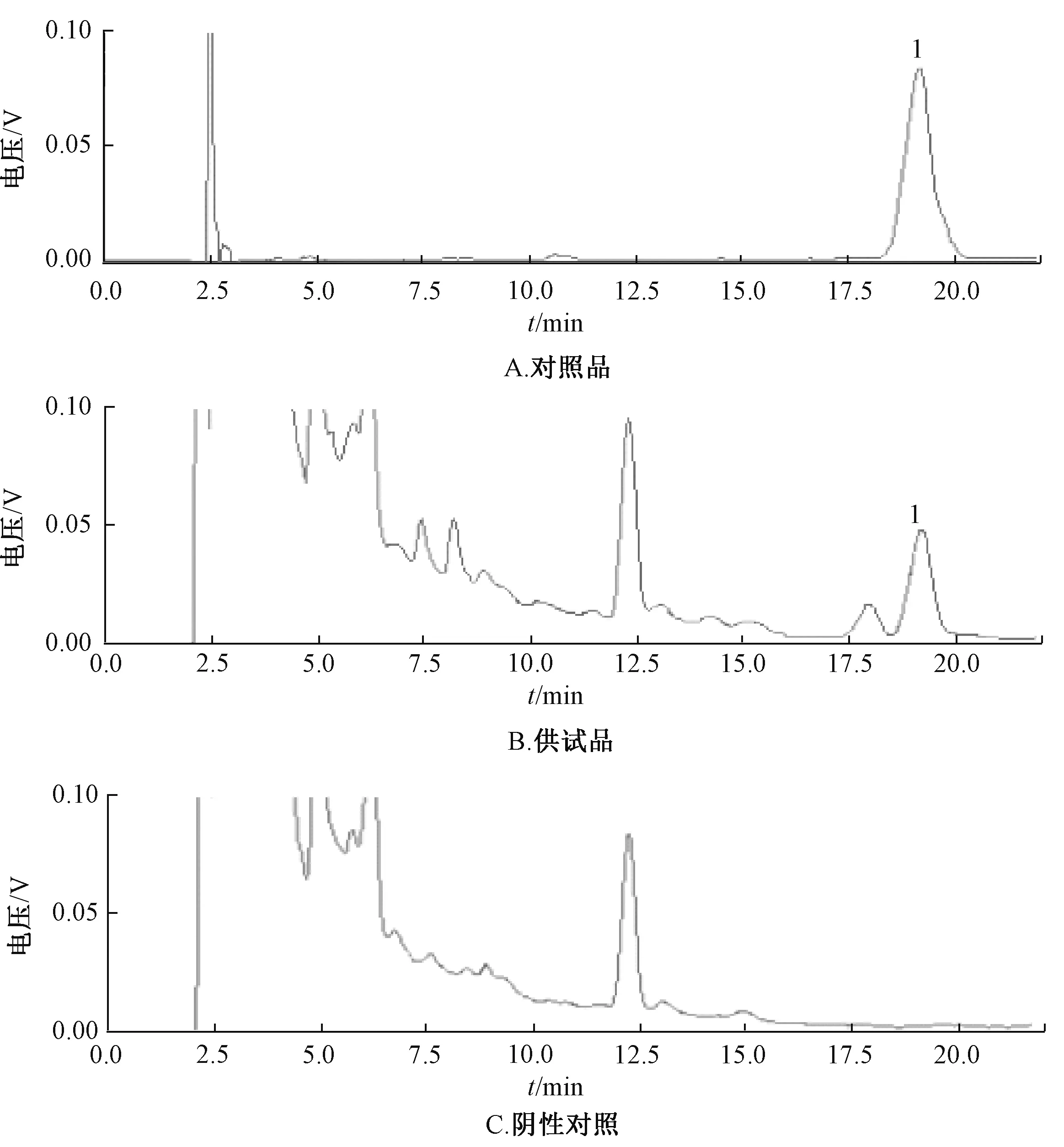
图4 熊果酸HPLC 色谱图

表1 熊果酸加样回收率试验结果(n=6)
2.3.9 样品含有量测定 取3 种不同来源的本品, 每种3份, 按“2.3.2” 项下方法制备供试品溶液, 在“2.3.3”项色谱条件下测定, 结果见表2。
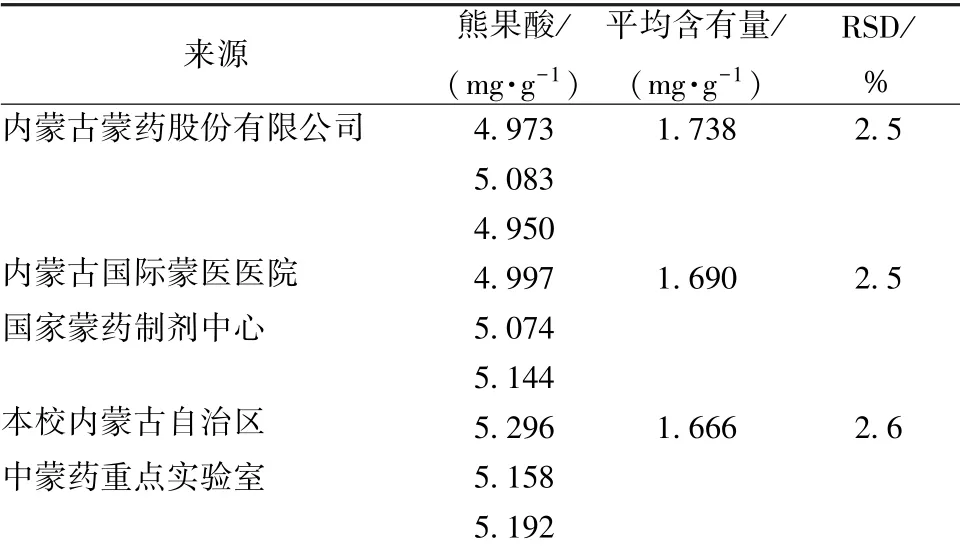
表2 熊果酸含有量测定结果(n=3)
3 讨论
3.1 显色剂筛选 文献报道可用10%香草醛硫酸溶液[12],但本实验发现显色效果不好, 层析斑点模糊不清。 再用10%硫酸乙醇作为显色剂, 显色效果较好, 得到了斑点清晰的薄层色谱。
3.2 流动相筛选 根据文献[13-14], 首先选择的流动相分别为乙腈-甲醇-水(60 ∶14 ∶27) 和乙腈-甲醇-水-醋酸铵(60 ∶14 ∶28 ∶0.6), 结果表明醋酸铵的存在对分离熊果酸和齐墩果酸至关重要, 能较好地分离两者。 经反复实验, 最终确定流动相为乙腈-甲醇-水-醋酸铵(60 ∶14 ∶30 ∶0.6),既可确保柱效的稳定性, 又能达到较好的分离效果。
3.3 保留时间考察 文献报道的保留时间是17 min[15],但本实验发现此时熊果酸、 齐墩果酸分离效果不理想, 故将其延长至21 min, 从而使两者得到了较好的分离。
猜你喜欢
中草药(2022年11期)2022-05-31
小天使·五年级语数英综合(2022年5期)2022-05-28
皮肤病与性病(2021年3期)2021-07-30
健康博览(2021年4期)2021-04-23
河南大学学报(医学版)(2020年4期)2020-08-26
世界科学技术-中医药现代化(2020年2期)2020-07-25
特别健康·下半月(2019年6期)2019-08-01
爱你(2019年25期)2019-07-16
恋爱婚姻家庭(2019年30期)2019-03-23
恋爱婚姻家庭·养生版(2019年10期)2019-02-25
