
一例交界型大疱性表皮松解症患儿LAMA3基因突变检测
2019-06-01 03:24孔祥生刘江波陈付英胡小华
中国麻风皮肤病杂志 2019年5期
孔祥生 刘江波 陈付英 胡小华 李 明
大疱性表皮松解症(epidermolysis bullosa,EB)是由于角质形成细胞或皮肤基底膜区内的蛋白结构异常导致的一类遗传性皮肤病,在新生儿中的发病率接近十万分之二[1]。其主要特点是患者皮肤和黏膜的脆性增加,即使是轻微的创伤也会引起水疱或大疱,在水疱破裂后出现表皮缺损和糜烂[2]。EB按照表皮的分离水平和水疱的形成位置可分为单纯型大疱性表皮松解症(epidermolysis bullosa simplex,EBS)、营养不良型大疱性表皮松解症(epidermolysis bullosa dystrophica,DEB)、交界型大疱性表皮松解症(epidermolysis bullosa junctional,JEB)和Kindler综合征四种主要类型[3,4]。其中EBS的水疱形成在表皮的基底细胞层,大多与编码角蛋白的基因KRT5和KRT14发生突变有关,少数与PLEC、DST和KLHL24基因突变有关[5,6];DEB的水疱形成在锚原纤维水平,与编码Ⅶ型胶原蛋白的基因COL7A1发生突变有关;JEB的水疱形成在基底膜带水平的透明板处,与编码层粘连蛋白-332的3个多肽(α3、β3和γ2)的基因LAMA3、LAMB3和LAMC2,编码α6β4整合素的基因ITGA6和ITGB4,编码大疱性类天疱疮抗原BPAG2的基因COL17A1发生突变有关[2,4,7];而Kindler综合征的水疱可以形成在多个位置,与FERMT1基因突变有关[3,8]。本研究通过靶向捕获-高通量测序,结合Sanger测序检测一例交界型大疱性表皮松解症患儿。
1 材料与方法
1.1 临床资料 患儿,男,1岁。出生时指(趾)甲显著肥厚,大约6个月时颈后、耳廓、腋后、臀部等易摩擦部位开始出现浆液性或血性水疱,部分水疱糜烂结痂(图1),皮损进行性加重,同时伴有声音嘶哑。患儿父母非近亲结婚,双方临床表型均未见异常。
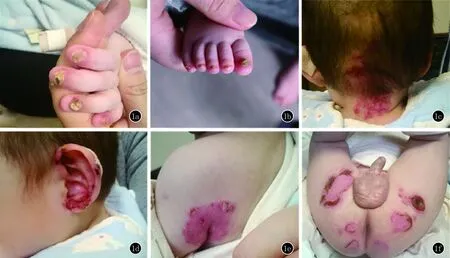
图1a、b:患儿指(趾)甲肥厚;c~f:患儿颈后、耳廓、腋后、臀部水疱糜烂结痂
1.2 核酸提取 在取得患儿父母的知情同意后用5 mL EDTA抗凝采血管分别取患儿及患儿父母的外周血2 mL,用AxyPrep血基因组DNA小量制备试剂盒(AP-MN-BL-GDNA-250G)提取基因组DNA;分别取患儿父母的头发20根,将毛囊部分剪到1.5 mL离心管中,各加入1 mL trizol,用天根公司RNAsimple Total RNA Kit(DP419)提取RNA。
1.3 文库构建 取1 μg患儿基因组DNA用covaris超声波破碎仪打断至片段长度在150~250 bp,用罗氏公司KAPA High Throughput Library Preparation Kit(7138008001)将DNA片段经末端补平、加A尾、连接接头和PCR富集得到DNA文库。然后将DNA文库与探针(该探针为安百隆生物公司设计的遗传性皮肤病检测专用panel,由罗氏公司合成)杂交过夜,在包被有链霉亲和素的磁珠帮助下将panel所涵盖的与遗传性皮肤病密切相关的541个基因的全部外显子捕获下来,经PCR富集得到捕获文库。
1.4 高通量测序及分析 将质检合格的捕获文库用Illumina Hiseq Xten高通量测序仪测序,然后使用BWA、Samtools和Picard软件将测序得到的reads比对到人类参考基因组GRCh37/hg19,生成的bam文件采用GATK系列软件进行局部重新比对,去除重复序列后检出变异,最后使用Annovar软件对vcf变异文件进行注释。以在ExAC_EAS及千人基因组数据库中突变频率小于0.01为标准进行筛选,结合患儿临床表型分析,在LAMA3基因上找到2个杂合突变。
1.5 DNA验证 使用NCBI网站上的Primer-blast设计DNA引物,F1:5’-GTCTGAAGTGACCAAGGATT-3’和R1:5’-CCCATATCTAGGGCAAGTTC-3’,F2:5’-TACAGGTTTTTCTTGGCACT-3’和R2:5’-AGTTAGCACTGAAGTCAAGG-3’,验证患儿2个突变的来源。PCR反应体系为20 μL,其中DNA模板50 ng,上、下游引物各0.5 μL(10 pM),2×Ex Taq premix(Takara)10 μL。PCR反应在ABI Veriti PCR仪上进行:预变性96℃ 3 min,变性96℃ 30 s,退火60℃ 30 s,延伸72℃ 1 min,35个循环,最后72℃延伸5 min。PCR产物用2%琼脂糖凝胶电泳,割胶回收后用ABI 3730XL型全自动测序仪测序(安百隆生物)。
1.6 cDNA验证 取正常人和患儿父母的毛囊RNA各800 ng,分别用Takara公司的PrimeScript RT Master Mix(RR036A)反转录得到cDNA。使用NCBI网站上的Primer-blast设计cDNA引物,F3:5’-TTGGGAGCCATTCAGAGACA-3’和R3:5’-TTCCTTTATCACTGTCTGGAGG-3’,F4:5’-CTTGGCTCACTCTGTATTGT-3’和R4:5’-CTACTGTCTGTGTCCAGTTC-3’,验证2个突变对RNA剪接的影响。PCR体系及扩增条件与DNA验证中相同,PCR产物用3%琼脂糖凝胶电泳,割胶回收后用ABI3730XL型全自动测序仪测序。
2 结果
通过靶向捕获-高通量测序在JEB的致病基因LAMA3(NM_001127718.2)上找到2个影响RNA剪接的杂合突变:exon12: c.1478+5G>A和exon34:c.4647+5G>C。DNA验证结果表明患儿的这2个突变分别遗传自其父亲和母亲(图2)。cDNA验证结果表明c.1478+5G>A导致mRNA上发生了缺失(图3a),即c.1381_1478del,原因是12号外显子3’端剪接位点前移了98bp(图4a),c.4647+5G>C导致mRNA上发生了插入(图3b),即c.4647_4648ins74,原因是34号外显子3’端剪接位点后移了74 bp(图4b),即插入序列为c.4647+1_4647+74。检索HGMD数据库(http://www.hgmd.org)、LOVD数据库(http://www.lovd.nl/LAMA3)以及最新LAMA3基因相关文献均未见有关以上两个突变的报道。
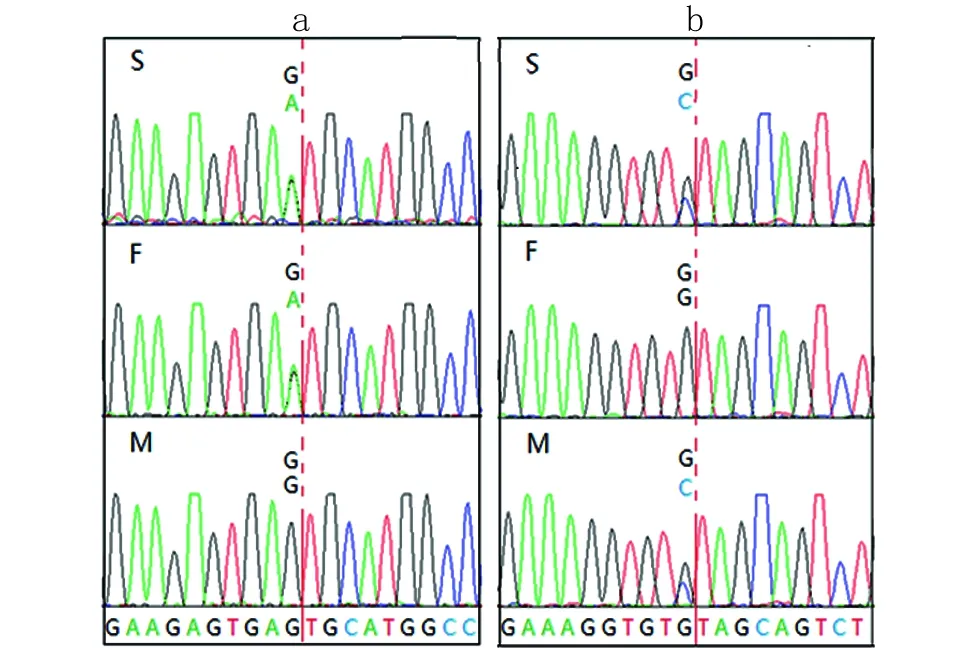
图2a:患儿(S)c.1478+5G>A遗传自其父亲(F);b:患儿(S)c.4647+5G>C遗传自其母亲(M)
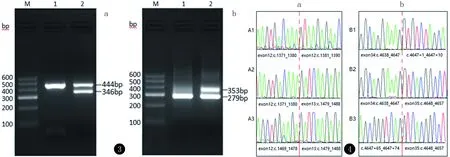
图3a:患儿父亲(2泳道)与正常人(1泳道)相比,mRNA上有缺失;b:患儿母亲(2泳道)与正常人(1泳道)相比,mRNA上有插入图4A1和A3为图3a中444bp条带测序结果,A2为图3a中346bp条带测序结果;B1和B3为图3b中353bp条带测序结果,B2为图3b中279bp条带测序结果
3 讨论
交界型大疱性表皮松解症与其他几种类型相比,不但基因杂合程度高,而且往往症状较重。特别是当LAMA3、LAMB3和LAMC2这三个基因中的任何一个发生突变时都可能导致层粘连蛋白-332的含量显著降低甚至消失,因而往往具有致死性[3,9,10],这种类型称为严重泛发型交界型大疱性表皮松解症。层粘连蛋白-332局限于鳞状上皮,如皮肤的基底膜中,它定位于锚丝并与半桥粒相连,对基底膜的构建和稳定具有非常重要的作用[11]。严重泛发型交界型大疱性表皮松解症十分罕见,在新生儿中的发病率在0.00004%~0.0004%[9],患儿常在出生时即有严重广泛性分布的大疱和大面积的皮肤剥脱,可于数日至数月内死亡,婴儿如幸存,其后仍可发生生长迟缓及中或重度的顽固性贫血,多数于2岁内死亡[12]。
本次检测到的LAMA3基因上的两个突变:c.1478+5G>A位于12号外显子的下游,导致在RNA剪接时12号外显子3’端的98bp外显子片段丢失,翻译时出现移码并在第469位形成提前终止密码子(p.Val461AspfsX9);c.4647+5G>C位于34号外显子下游,导致在RNA剪接时34号外显子下游的74 bp内含子片段被插入到34号和35号外显子之间,翻译时出现移码并在第1557位形成提前终止密码子(p.Thr1551CysfsX7),二者均导致LAMA3基因的编码蛋白错误和截短,直接引起其功能改变。
交界型大疱性表皮松解症预后差,至今无有效治疗方法,产前诊断是主要干预手段。我们应用靶向捕获-高通量测序技术在本例患儿中新发现2个影响RNA剪接的突变,增加了中国人群LAMA3基因的突变数据库,为该患儿的父母预防下一胎患病所需的产前诊断打下了基础,也为探讨该病的发病机制以及将来的基因治疗提供了参考。
猜你喜欢
电子科技大学学报(2022年5期)2022-10-29
卫星应用(2022年3期)2022-05-23
中国典型病例大全(2022年11期)2022-05-13
法人(2021年12期)2021-05-09
中国生殖健康(2020年4期)2021-01-18
透析与人工器官(2020年1期)2020-11-16
中华养生保健(2020年7期)2020-11-16
中国心脏起搏与心电生理杂志(2020年5期)2020-10-31
肿瘤预防与治疗(2019年6期)2019-07-30
中国心脏起搏与心电生理杂志(2019年1期)2019-03-02
