
基于PCR的两步法丝状真菌基因敲除方法
2019-05-28 02:04蒋君梅1勇1任明见
种子 2019年4期
谢 鑫, 蒋君梅1, 王 勇1, 任明见
(1.贵州大学农学院, 贵阳 550025; 2.国家小麦改良中心贵州分中心, 贵州 贵阳 550025)
真菌在自然界广泛分布,与人类生活的各个方面都有着密不可分的联系[1]。因此对真菌进行研究,尤其对真菌的基因功能进行研究具有重要意义。通过对真菌DNA进行遗传改造是基因功能研究的重要技术,目前,对丝状真菌进行遗传改造的方法主要有PEG介导的原生质体转化、农杆菌介导的转化(A.tumeafeiensmediatedtransformation, ATMT)、电击转化和基因枪转化等[2-3]。基因敲除是研究基因功能的手段之一,利用同源重组原理对感兴趣的基因进行敲除,通过观察敲除突变体的表型变化从而明确该基因的功能,最终实现改良或防治真菌的目的。目前,常用的基因敲除的方法有: 1) 同源重组基因敲除,即利用基因两侧的同源臂在位点特异性DNA重组酶的作用下进行基因敲除,该法是最普遍的基因敲除方法; 2) 基因沉默(RNA silence)诱导的基因敲除,即通过构建小的双链RNA片段干扰基因的表达,并且这种干扰效应会持续“传递”到子代细胞,从而造成基因敲除; 3) 转座子和逆转录转座子标签法,即通过转座子在基因组中 “跳跃”,从而造成染色体的畸变或基因的缺失[4-6]。以上的基因敲除方法最终都需要筛选标记对敲除突变体进行筛选,丝状真菌基因敲除时常用的筛选标记有潮霉素、氯嘧磺隆(SUR)和G 418等[7],存在筛选效率低、假阳性率高等缺点。因此,本研究对此做了改进,利用同源重组的原理,采用两步法PCR进行基因敲除,并利用潮霉素和绿色荧光蛋白(GFP)同时进行转化子的筛选,大大提高了筛选效率,其操作流程见图1。
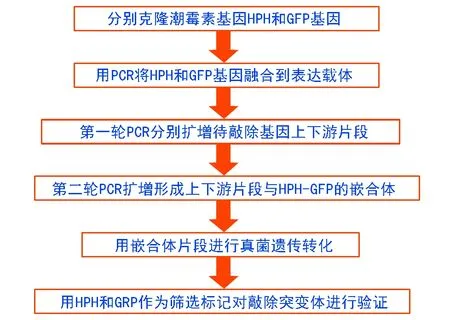
图1 本研究流程图
1 材料与方法
1.1 试验材料
1.1.1供试菌株
稻瘟菌菌株GUY 11野生型菌株来自贵州大学植物病理学教研室。
1.1.2供试试剂
燕麦、水解酪蛋白、酵母提取物、蔗糖、酵母提取物、琼脂粉、溶菌酶(sigma)PEG 8000(sigma)、Tris-HCl、氯化钙、氯化钠、山梨醇、FastPfu Fly高保真酶(全式金)、pEASY-Blunt克隆载体(全式金)、苯酚、氯仿、乙醇、CTAB、金刚砂(sigma)等。
1.1.3培养基
燕麦培养基(1 L):燕麦30 g(用料理机打碎),琼脂粉15 g,蒸馏水定容至1 L,121 ℃灭菌20 min。
完全培养基(CM):水解酪蛋白6 g,酵母提取物6 g,蔗糖10 g,蒸馏水定容至1 L,121 ℃灭菌20 min。
酶解液:0.1 g溶菌酶溶解并定容至10 mL 0.7 M NaCl溶液中,抽滤除菌。
1×STC(200 mL):山梨醇43.6 g,50 mM Tris-HCl, pH=8.0 10 mL,50 mM CaCl210 mL,蒸馏水定容至200 mL,121 ℃灭菌20 min。
40%PTC(100 mL):PEG 8000 4 g加1×STC定容至100 mL,抽滤除菌。
TB 3 培养基(1 L):酵母提取物6 g,水解酪蛋白6 g,蔗糖200 g,蒸馏水定容至1 L,121 ℃灭菌20 min。
1.1.4实验设备
本实验所涉及的主要仪器有:离心机、光学显微镜、光照培养箱、荧光显微镜、摇床、料理机等。
1.2 试验方法
1.2.1稻瘟菌培养
将稻瘟菌接种于新鲜的燕麦培养基,25 ℃光照培养8~10 d。
1.2.2PCR扩增、连接等分子生物学方法
用于载体构建的DNA 扩增所用的PCR 扩增体系如下:5×buffer 10μL, FastPfu Fly DNA Polymerase 1μL (全式金),2.5 mmol dNTPs 4μL,DNA 模板 0.5μL (约 5 ng 质粒 DNA),10μmol 上下游引物各1μL,ddH2O 32.5μL。
以下为PCR 扩增条件:95 ℃ 2 min,35 个循环(95 ℃ 20 s,58 ℃ 20 s,72 ℃ 0.5~1 min(根据DNA 片段长度确定),72 ℃ 5 min。PCR 产物经纯化后,连接到 pEASY 载体。采用1%琼脂糖对PCR产物进行检测。
HPH-GFP融合表达载体的构建采用如下方法进行:分别用TRP-F/HYG-GFP-R和HYG-GFP-F/GFP-T引物对从pBHt 2载体和PYBA 1132载体扩增带有启动子TrpC的潮霉素HPH片段和GFP片段,并以此PCR产物HPH和GFP片段为模版,用TRP-F/GFP-T引物扩增,获得融合的HPH-GFP片段,并将该片段插入全式金pEASY-Blunt克隆载体,挑单克隆DNA测序鉴定没有突变的克隆保存备用(图2)。
1.2.3稻瘟菌原生质体的制备及转化
原生质体制备:将新鲜的固体培养基上的菌丝切成小块,置于液体完全培养基中,28 ℃ 120 r·min-1培养3 d;过滤收集菌丝,用0.7 M NaCl溶液洗涤菌丝去除残余培养基;将菌丝转移至溶菌酶溶液中, 30 ℃ 90 r·min-1裂解3~4 h,镜检原生质体裂解情况,用 1×STC洗剂重悬原生质体至 5 × 107个·mL-1。
原生质体转化:采用40% PEG(PTC)对原生质体进行转化, 200μL原生质体与100μL PCR产物,孵育 20 min;加入PTC 1.5 mL,孵育20 min;最后加入TB 3培养基,28 ℃,90 r·min-1摇床上复苏12 h。
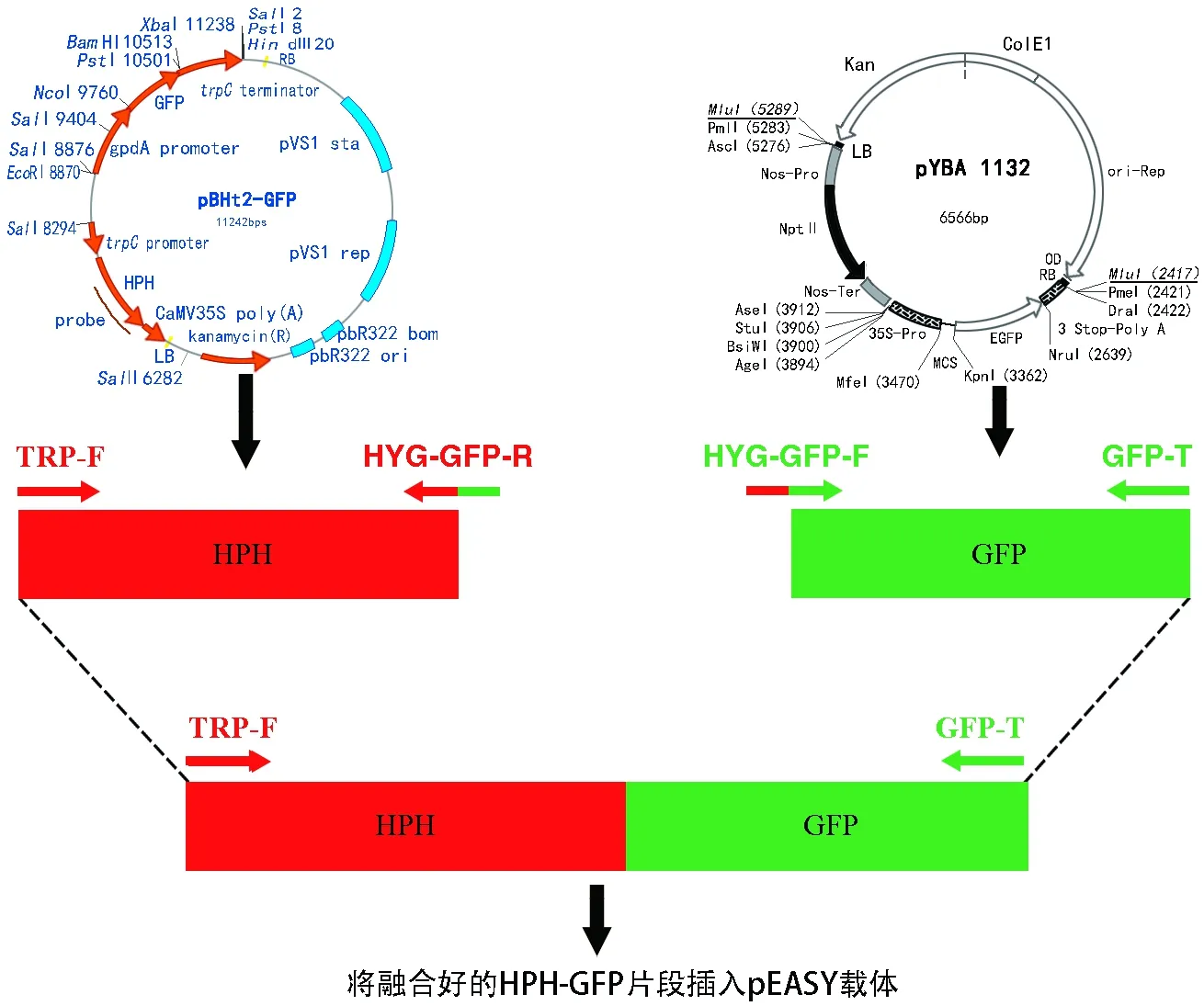
图2 HPH-GFP融合表达载体
表1 引物列表

引物名称 引物序列(5’-3’)TRP-FGCTGGAGCTAGTGGAGGTCAAHYG-GFP-RCTCGCCCTTGCTCACCATTTCCTTTGCCCTCGGACGAGTHYG-GFP-FACTCGTCCGAGGGCAAAGGAAATGGTGAGCAAGGGCGAGGFP-TTCACTTGTACAGCTCGTCCATM13-FTGTAAAACGACGGCCAGTM13-RCAGGAAACAGCTATGACCHY-FGCCGTGGTTGGCTTGTATGFP-RTCGCCGGACACGCTGAAMoNACα_UAAGGAATCTTTGGGACTMoNACα_AFTATCCAATCGGCATTAGGGMoNACα_ARACTGGCCGTCGTTTTACATTTCGCGGGTGGTCAAMoNACα_BFGGTCATAGCTGTTTCCTGACACTCGGTCTTGGTTTATTGMoNACα_BRTGAGGACGGAGCGGTAGMoNACα_NFGACCGCCGTCATCCACTMoNACα_NRTCACCGAAGACGCTGTAATGMoNACα_DBCCTGGCAGTCTCAAT
1.2.4稻瘟菌DNA的提取
采用微量法提取法对稻瘟菌DNA进行提取,方法如下:在1.5 mL离心管中 加入约100μL 金刚砂,加入400μL CTAB提取buffer(CTAB 10 g,Tris 6.06 g,EDTA 1.46 g,NaCl 0.5 g,加水定容到500 mL),用灭菌的棉签刮取适量菌丝放入管中,加入400μL的苯酚∶氯仿∶异戊醇(25∶24∶1),把离心管置于37 ℃摇床250 r·min-1摇1 h,13 000 r·min-15 min,吸取250μL 上清液,加入800μL 无水乙醇,-20 ℃沉淀30 min,70%乙醇洗1次,室温晾干,加入50μL ddH2O溶解DNA,取1μL进行PCR。
1.2.5引 物
本研究所用引物见表1。
2 结果与分析
本实验包括两轮PCR,其具体操作流程如图3所示。
2.1 第一轮PCR扩增
分别用HYG-GFP-F/GFP-T和TRP-F/HYG-GFP-R引物对从PYBA 1132载体和pBHt 2载体扩增GFP片段(720 bp)和带有启动子TrpC的潮霉素HPH片段(1 352 bp)(图4泳道2和3)。然后用M 13-F/M 13-R引物从HPH-GFP融合表达载体上克隆出HPH-GFP片段,备用(图4泳道4)。分别用基因上、下游片段特异引物AF/AR和BF/BR对基因组DNA进行扩增,其中AR引物和BF引物5’端分别加ACTGGCCGTCGTTTTACA和GGTCATAGCTGTTTCCTG接头序列,得到基因上下游特异片段(图3)。
2.2 第二轮PCR扩增
分别用第一轮PCR所得到的HPH-GFP片段和基因上下游特异片段为模版,以引物AF/GFP-R和HY-F/BR扩增,得到2个上下游片段 HPH-GFP的嵌合体片段,1∶1混合2个片段(图3),然后用嵌合体片段进行真菌遗传转化。
2.3 稻瘟菌NACα基因敲除
为检验本研究方法的可行性,用本方法对稻瘟菌NACα基因进行敲除,NACα基因序列来自于稻瘟菌数据库(http://fungidb.org/fungidb/),具体步骤如下:
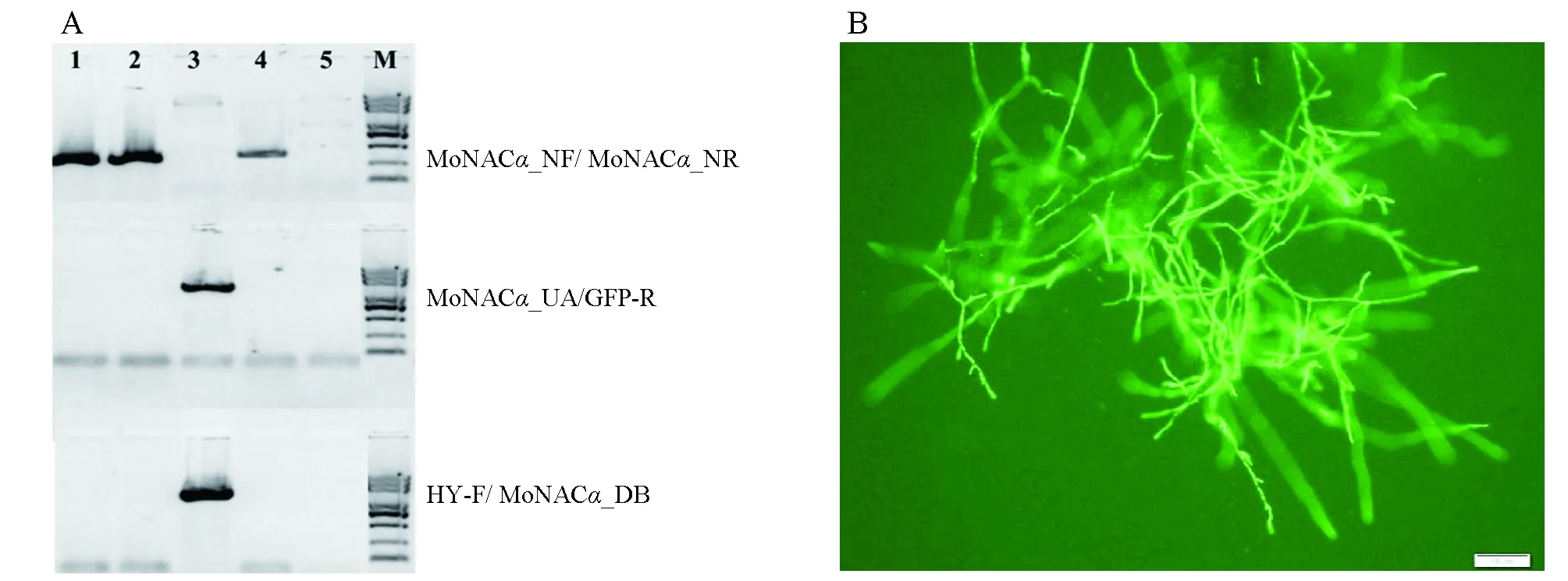
注:A为转化子PCR鉴定;1,2为泳道为野生型基因组对照;3,4为转化子基因组DNA;5为水对照。B为转化子荧光鉴定。图5 转化子的鉴定
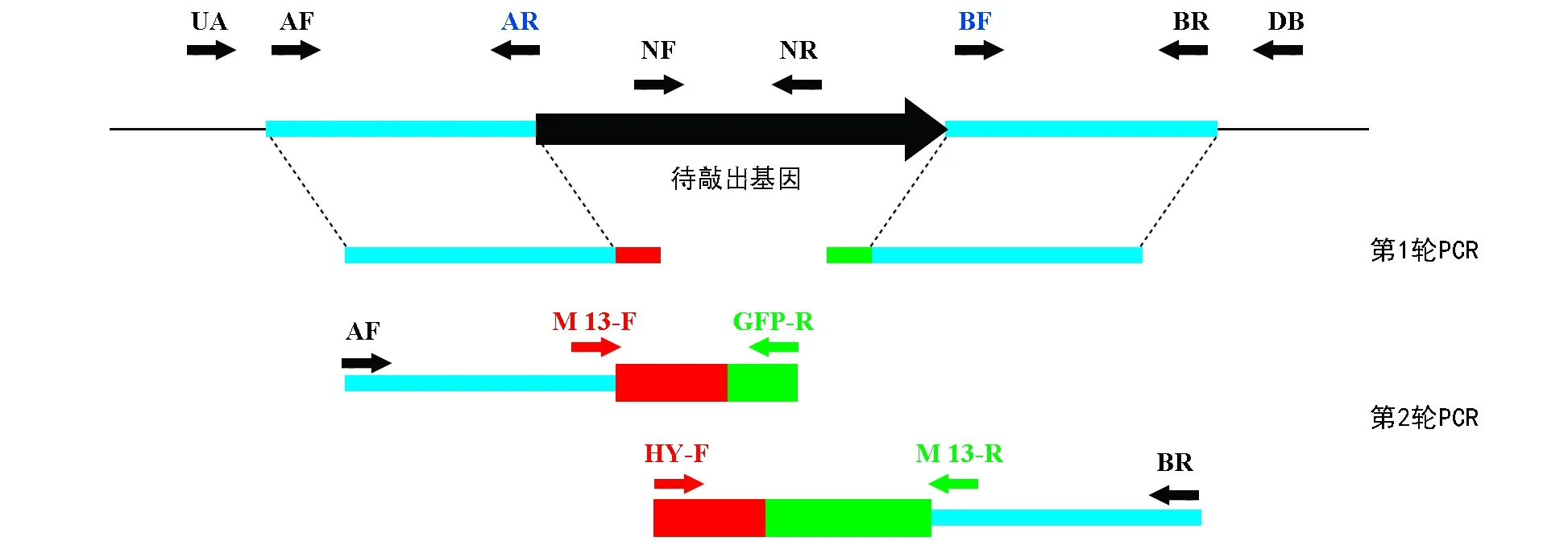
图3 两步PCR法构建基因敲除流程图
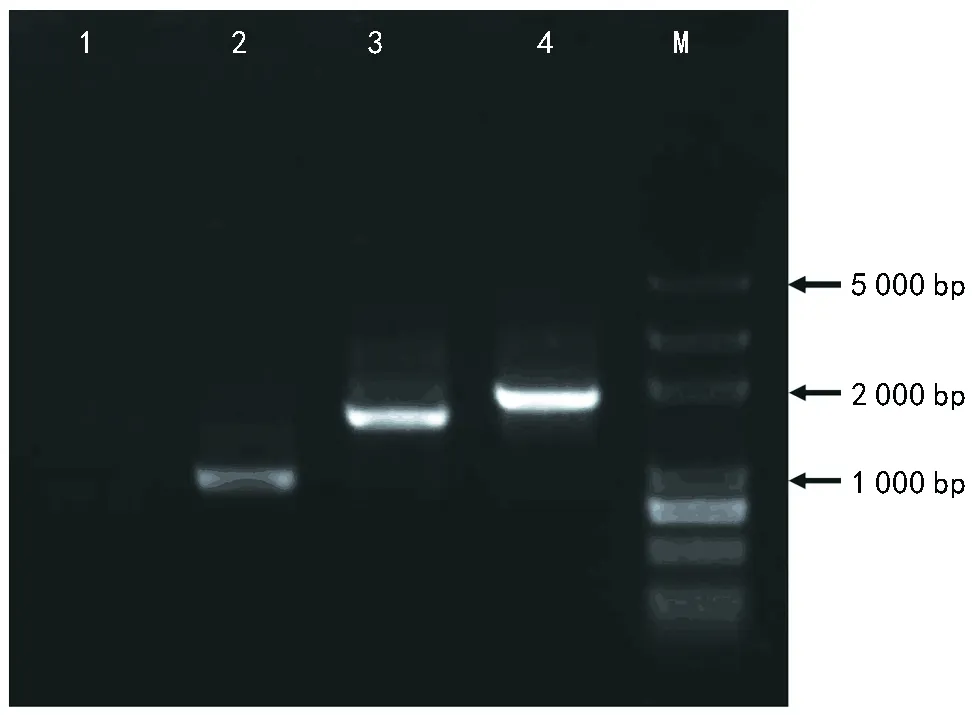
注:1为泳道为阴性对照;2为泳道为GFP片段;3为泳道为HPH片段;4为泳道为HPH-GFP片段;M为Marker。图4 HPH-GFP融合片段PCR扩增结果
1) 经过第一轮PCR分别扩增MoNACα基因上下游片段:分别用引物MoNACα_AF/ MoNACα_AR和MoNACα_BF/ MoNACα_BR从稻瘟菌基因组中扩增基因上游片段(595 bp)和下游片段(550 bp);
2) 第二轮PCR扩增形成MoNACα基因上下游片段与HPH-GFP的嵌合体:分别用从第一轮PCR得到的上下游产物和HPH-GFP片段为模版,以引物AF/GFP-R和HY-F/BR扩增,得到2个上下游片段-HPH-GFP的嵌合体片段,1∶1混合2个片段,备用;
3) 原生质体转化:将混合的片段转入稻瘟菌原生质体进行转化,25 ℃培养1周,挑取转化子进行鉴定。
2.4 转化子鉴定
1) 转化子DNA的PCR验证:用CTAB法提取转化子DNA,并分别用MoNACα_UA/GFP-R、HY-F/ MoNACα_DB和MoNACα_NF/ MoNACα_NR这3对引物扩增提取的转化子DNA来验证是否为阳性转化子。结果显示阳性转化子用MoNACα_UA/GFP-R、HY-F/ MoNACα_DB引物可以扩增出条带(图5第3泳道),而用引物MoNACα_NF/ MoNACα_NR则不能扩增出条带(图5 A第3泳道),相反野生型对照用MoNACα_NF/MoNACα_NR可以扩增出条带(图5 A第1,2泳道),说明第3泳道的转化子为阳性,其MoNACα基因被敲除,而第4泳道的转化子则为未敲除的阴性转化子。
2) 用荧光显微镜观察转化子是否有GFP荧光:为进一步确定是否敲除转化子是否为阳性,用牙签挑取阳性转化子少许菌丝,打散到加水的载玻片上,在荧光显微镜下观察转化子是否有GFP信号,结果显示阳性转化子在荧光显微镜下有GFP信号(图5 B),用荧光观察的方法与PCR鉴定方法结果一致。由于用荧光鉴定免去了提取真菌DNA复杂繁琐的程序,因而采用本研究进行突变体鉴定更加高效而简单。
3 结论与讨论
随着功能基因组学时代的到来,对基因的功能研究已经变得越来越普遍。在丝状真菌的研究中,通常采用同源重组的方法进行基因敲除,然后采用抗生素等筛选标记对转化子进行筛选,最后采用提取转化子的DNA,用PCR对转化子进行鉴定[8]。转化子鉴定工作复杂而繁琐,对于动辄成百上千个转化子鉴定,一一提取DNA,PCR鉴定费时费力费钱,虽然也有研究报道简化的DNA提取方法,起到了减少工作量的目的,但还远远不能满足快速鉴定的目的。因此,本研究采用GFP荧光标记方法进行检测,使得转化子鉴定工作效率大大提高,同时也减少了酚氯仿等试剂的使用,安全性也大大提高。
有研究采用三轮PCR的方法,形成基因敲除嵌合体转化镰刀菌获得基因敲除突变体[9],本研究采用两步法PCR的方法进行基因敲除,步骤进一步简化,有利于大规模的基因敲除工作。转化子的筛选通常依赖标记抗生素的筛选,也有报道采用失活聚酮合酶基因的方法作为标记基因(该基因失活后会产生白化现象)[10],这可以作为今后借鉴的思路。
猜你喜欢
食品安全导刊(2021年21期)2021-08-30
福建农业学报(2021年2期)2021-05-31
新农业(2020年21期)2020-11-19
数学大王·低年级(2020年8期)2020-08-14
西南农业学报(2019年6期)2019-07-18
大自然探索(2019年1期)2019-01-24
大自然探索(2017年3期)2017-04-07
小雪花·初中高分作文(2016年9期)2016-05-14
营销界(2015年23期)2015-02-28
食品工业科技(2014年23期)2014-03-11
