
还原-氧化预处理对Co3O4催化分解N2O性能的影响
2019-04-18 08:57王永钊胡晓波武瑞芳刘晓丽赵永祥
燃料化学学报 2019年4期
郑 珂, 王永钊, 胡晓波, 武瑞芳, 刘晓丽, 赵永祥
(1. 精细化学品教育部工程研究中心, 山西 太原 030006; 2. 山西大学 化学化工学院, 山西 太原 030006)
N2O作为一种空气污染物近年来受到越来越多的关注。N2O具有强烈的温室效应,其全球变暖潜能值分别是CO2和CH4的310倍和21倍[1],同时N2O会对臭氧层造成破坏,且可在平流层中稳定存在约150年。N2O主要来源于硝酸、己二酸的生产过程及汽车尾气的排放。目前,大气中N2O浓度正以每年0.2%-0.3%的速率增加[2]。因此,消除N2O对于保护生态环境十分重要且迫在眉睫。
消除N2O的方法主要有三种:热分解法、选择性催化还原和催化分解法[3]。其中,催化分解法因其简单高效而成为研究和应用的首选。催化分解催化剂包括三类:贵金属催化剂[4-6]、金属氧化物催化剂[7-10]和分子筛催化剂[11]。其中,金属氧化物催化剂具有良好的化学稳定性和热稳定性,且成本低廉,受到研究者的青睐[12]。
在各种金属氧化物催化剂中,尖晶石型氧化物(AB2O4),由于结构中含有两种不同价态的金属离子,可有效形成氧化还原对,表现出良好的催化性能[13]。Co3O4作为一种典型的尖晶石型氧化物,不仅含有Co2+↔Co3+氧化还原对,而且具有较弱的Co-O键键能,在低温下易形成氧空位[14],因而在CO氧化、费-托合成以及N2O分解等反应中显示出优异的催化活性[15-17]。为了进一步提高Co3O4催化N2O分解的活性,以及对杂质气体(H2O、O2等)的耐受性,人们一般通过掺杂二价或三价金属离子对Co3O4尖晶石结构改性,提高其氧化还原能力[18-21]。此外,有研究表明,对催化剂进行不同的预处理也可改善其催化性能[21]。Sadykov等[22]发现,Co3O4在He气氛下预处理会发生表面重建,使CO氧化活性增加。Yu等[23]研究了不同预处理条件对Co3O4催化CO氧化活性的影响,结果表明,经还原-氧化预处理或在惰性气氛下干燥可引起表面氧空位数量增多,导致催化活性增强。Yang等[24]同样考察了还原-氧化预处理对Co3O4/Al2O3催化剂CO氧化活性的影响,发现预处理后催化剂晶粒粒径减小,同时活性中心(Co3+)数量增加。Hussain等[25]则采用还原-氧化-还原多步预处理方式,提高了Co在载体中的分散度和催化剂的比表面积,从而促进了其费-托合成的催化性能。Cai等[26]发现,20%Co/SiO2催化剂经还原-氧化-还原预处理后,Co粒子粒径减小,同时指出金属颗粒的再分散发生在氧化过程。王俊刚等[27]研究了还原-氧化预处理对Co/MCM-41分子筛催化剂费-托合成性能的影响,发现预处理促进了Co物种在载体上的分散,且降低了还原温度,同时增强了活性物种与载体间相互作用,提高了对C5-11选择性。可见,不同预处理方式对Co3O4以及负载型Co3O4催化剂的CO氧化、费-托合成催化性能均产生了积极的影响。但目前关于预处理对Co3O4催化分解N2O性能的影响鲜有报道。
本研究采用液相沉淀法制备了Co3O4催化剂,并对其进行还原-氧化预处理。考察了预处理前后催化剂催化分解N2O性能。采用XRD、Raman、H2-TPR、XPS和O2-TPD等对催化剂进行表征,并与其催化性能相关联。
1 实验部分
1.1 实验原料
Co(NO3)2·6H2O,分析纯,天津市大茂化学试剂厂;Na2CO3,分析纯,天津市凯通化学试剂有限公司。
1.2 催化剂的制备
采用液相沉淀法制备Co3O4催化剂。将化学计量的Co(NO3)2·6H2O溶解在去离子水中,在室温下将Na2CO3(1 mol/L)溶液缓慢滴入混合溶液中直至pH值达到9。将所得悬浮液搅拌1 h并老化3 h。过滤悬浮液并用去离子水洗涤直至滤液pH值达到7。最后,将沉淀物在120 ℃下干燥12 h,在400 ℃下空气中焙烧3 h。所得催化剂标记为Co3O4。
催化剂的还原-氧化预处理:将新鲜催化剂装入管式炉中,通入体积分数5% H2/N2混合气,调节流量为20 mL/min,以3.5 ℃/min的速率升温至350 ℃,恒温4 h后,自然冷却至室温,切换为N2,以20 mL/min流量吹扫30 min,然后再切换为空气,流量为20 mL/min,并以相同的速率升温至250 ℃,恒温4 h,然后降至室温。所得预处理后的催化剂标记为Co3O4-RO。
1.3 催化剂的表征
氮气物理吸附-脱附在Micromeritics ASAP-2020仪器上进行。催化剂预先在150 ℃真空条件下脱气预处理 5 h,然后在液氮温度下进行N2吸附-脱附。比表面积(ABET)用BET程序测定,平均孔径和孔体积通过BJH方法测定。
粉末X射线衍射谱图采用Bruker D8 Advance X-ray衍射仪测量,使用CuKα辐射(λ= 0.154 nm),10°-80°扫描,扫描速率为 2.4(°)/min。
拉曼光谱用Horiba Soiontifc LabRam HR Erolution Raman光谱仪记录,激发波长为532 nm,室温下功率为10 MW。光谱为100-800 cm-1,分辨率为1 cm-1。
采用Micromeritics AutoChemII 2920分析仪进行H2-TPR表征。将40-60目催化剂 30 mg置于反应管,在体积分数5%H2/N2(30 mL/min)中以10 ℃/min从室温升至600 ℃。通过热导检测器(TCD)测量H2消耗量,并通过CuO定量还原为金属铜来校准。
X射线光电子能谱在ESCALAB 250Xi光谱仪上进行测量,使用AlKα辐射作为激发源,结合能(EB)以碳的C 1s结合能284.8 eV为基准进行校正。
O2-TPD实验使用Micromeritics AutoChemII 2920化学吸附分析仪进行测试。将催化剂(300 mg)在流动的O2(30 mL/min)气氛中300 ℃下预处理1 h,然后冷却至50 ℃以下。切换为He(30 mL/min)吹扫,待基线平稳后,以5 ℃/min的速率从50 ℃升温至400 ℃,同时采用TCD检测O2脱附信号。
1.4 催化剂的活性评价
N2O催化分解在连续流动微反应装置上进行。将300 mg催化剂(40-60目)置于反应器中,热电偶放入催化剂床层以测量温度。将体积分数0.1% N2O/Ar气体混合物通过催化剂,空速(GHSV)为10000 h-1。通过配有热导检测器的Agilent 7890B气相色谱仪分析气体组成。测试在100-500 ℃进行,升温速率为5 ℃/min。催化活性由N2O转化率表示,其通过以下公式计算:
x=([N2O]in-[N2O]out)/[N2O]in×100%
(1)
式中,[N2O]in表示反应前的N2O浓度,[N2O]out表示反应后的N2O浓度。
为了进一步研究杂质气体对N2O催化分解活性的影响,在原料气中分别引入体积分数2.3%H2O或体积分数2%O2进行活性评价。
根据以下等式计算比反应速率(r)、速率常数(k)和活化能(Ea)[20]:
(2)
(3)
(4)
式中,p0是大气压(101 kPa),F(mL/s)是进料气体混合物的总流量,R是气体常数(8.314 J·K-1·mol-1),mcat(g)是催化剂的质量,T0是室温(298 K),c(mol/m3)是反应器出口的N2O浓度,T(K)是反应温度。
2 结果与讨论
2.1 催化剂的XRD表征
图1为预处理前后催化剂的XRD谱图。由图1可知,两种催化剂均在19.6°、31.7°、36.9°、44.8°、59.7°和65.2°处出现了衍射峰,分别对应于Co3O4尖晶石结构的(111)、(220)、(311)、(400)、(511)和(440)晶面(JCPDS 43-1003)[18]。与Co3O4相比,Co3O4-RO催化剂的衍射峰强度明显降低且峰略微变宽,尤其是59.7°和65.2°的衍射峰消失,表明Co3O4-RO的结晶度变差,晶粒粒径减小,这是由于还原-氧化预处理过程中原有的Co3O4尖晶石结构被破坏,并进行结构重构所致。利用谢乐方程计算,结果显示Co3O4的平均晶粒粒径为24.5 nm,而Co3O4-RO的平均粒径骤降至8.1 nm。很显然,对于Co3O4而言,还原-氧化预处理可有效降低其结晶度和晶粒粒径,这不仅与文献报道一致[24],同时也与Co3O4中掺杂Sn、Ag等第二组分所起到的效果类似[17,18]。此外,表1中显示Co3O4的晶胞参数为0.8084 nm,还原-氧化预处理后则明显变大,这也表明由于结构重构导致Co3O4-RO结晶度变差[28]。由表1还可知,经还原-氧化预处理后Co3O4-RO的比表面积显著减小。
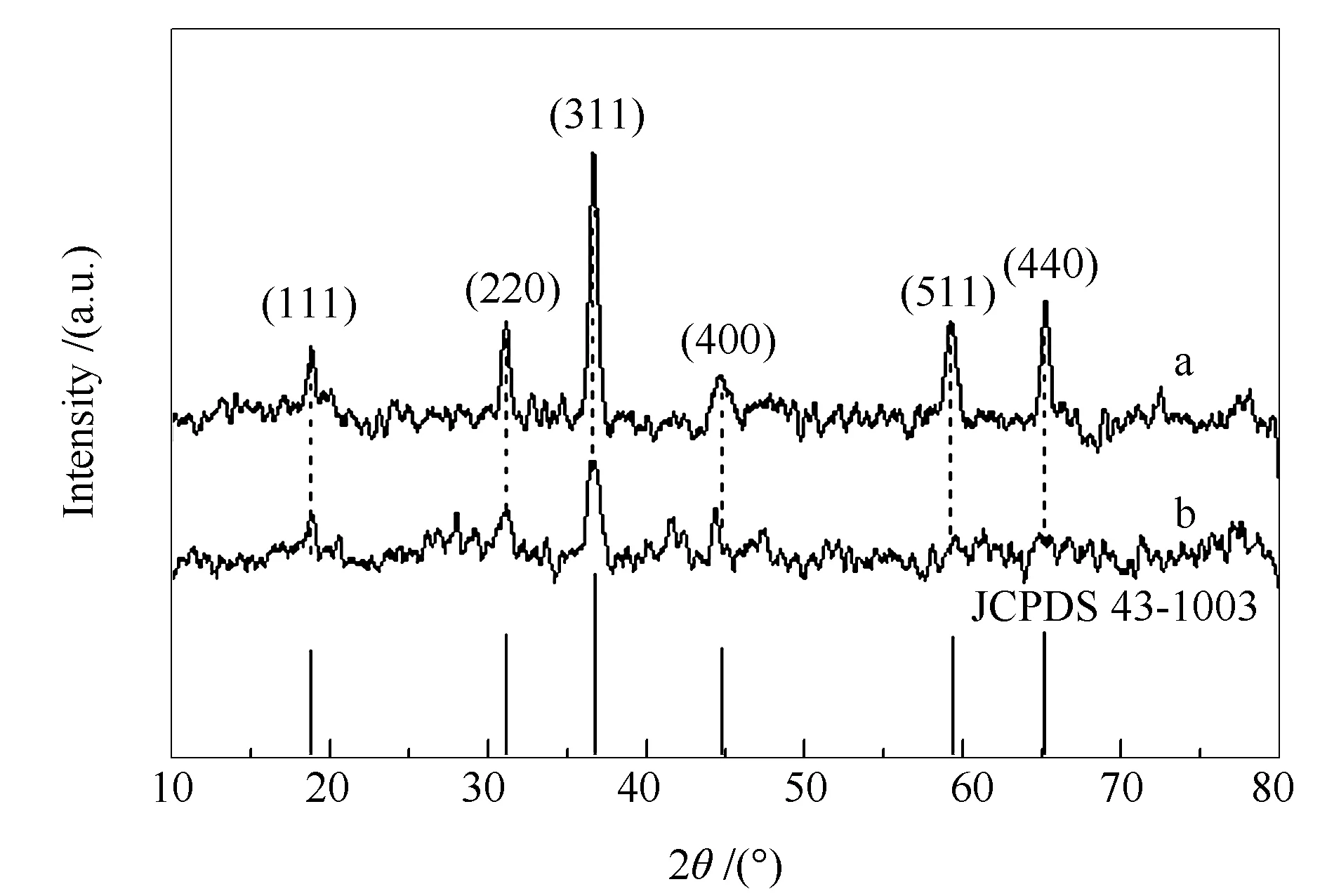
图 1 Co3O4和Co3O4-RO催化剂的XRD谱图

表 1 Co3O4和Co3O4-RO催化剂的织构性质和晶粒粒径
2.2 催化剂Raman表征
图2为两种催化剂的Raman谱图。由图2可知,Co3O4催化剂在195、461、503、602和664 cm-1处有五个拉曼峰,分别归属为Co3O4的3×F2g、Eg和A1g振动模式。Co3O4属于空间点群Oh、Fd3m[29],A1g对应于O7h对称中的八面体位点CoO6的振动,Eg和F2g则对应于四面体位点CoO4的振动[27]。与Co3O4催化剂相比,经还原-氧化处理后的Co3O4-RO催化剂虽然也在相同的位置出现了拉曼峰,但峰型相对宽化。以664 cm-1处的最强峰为例,Co3O4和Co3O4-RO的半峰宽分别为27和31 cm-1,进一步表明经还原-氧化处理后Co3O4-RO结晶度降低,这与XRD表征结果一致。
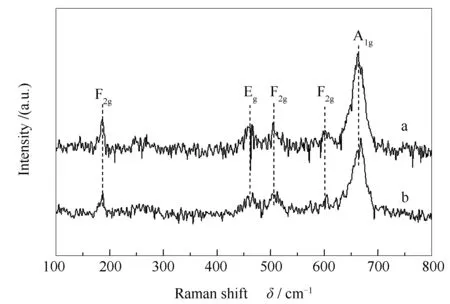
图 2 Co3O4和Co3O4-RO催化剂的Raman谱图
2.3 催化剂氧化还原性表征
图3为两种催化剂的H2-TPR谱图。由图3可知,Co3O4和Co3O4-RO在测试温度范围内均有两个耗氢峰。250-350 ℃的第一个峰归属为Co3O4中Co3+到Co2+的还原,350-450 ℃的第二个峰归属为Co3O4中Co3+还原生成的Co2+和结构中原有的Co2+共同到Co0的还原[30]。对于Co3O4而言,两个还原峰的峰顶温度分别为331和409 ℃,而Co3O4-RO的还原温度均向低温方向移动,尤其是Co2+到Co0的还原峰,峰顶温度从409 ℃降至398 ℃,表明经还原-氧化预处理后催化剂的氧化还原能力显著增强。一般来说[31],金属氧化物的分散度越高,晶粒粒径越小,其H2-TPR的还原峰温度越低。根据XRD表征结果可知,Co3O4-RO的平均晶粒粒径小于Co3O4,因此,Co3O4-RO的还原温度向低温处偏移。同时,还原温度降低表明,Co3O4-RO中Co-O键更易于断裂。可见,还原-氧化预处理在使催化剂发生结构重构的同时,还削弱了Co-O键。

图 3 Co3O4和Co3O4-RO催化剂的H2-TPR谱图
由还原峰面积计算的氢气消耗量见表2。由表2可知,Co3O4催化剂峰β/峰α的氢气消耗量比约为2.96,这与理论值相接近(3.0)。然而,对于Co3O4-RO而言,峰β/峰α之比仅为2.29,明显低于Co3O4。这表明还原-氧化预处理造成结构重构的同时也引起了催化剂中Co3+/Co2+的变化。

表 2 Co3O4和Co3O4-RO催化剂的H2消耗量
2.4 催化剂XPS表征
图4(a)为Co3O4和Co3O4-RO的Co 2pXPS谱图。由图4(a)可知,两种催化剂均在778.5-782.0和793.5-796.0 eV处出现特征峰,两峰之间的差值(ΔE)为Co 2p自旋轨道分裂能。ΔE为(15±0.1) eV(见表3),表明Co物种的存在形式为尖晶石型Co3O4[32],这与XRD表征结果一致。对于Co3O4,778.5-782.0 eV处特征峰可分峰拟合为781.1和779.4 eV两个峰,分别归属为Co2+(n)和Co3+(m),同时789.6 和788.2 eV处的卫星峰(u)也标志着Co2+的存在[33]。经还原-氧化预处理后,Co3O4-RO的Co 2p3/2峰略微向低结合能处位移,表明Co周围电子云密度降低,其可能会削弱Co-O键。
图4(b)为两种催化剂O 1s的XPS谱图。经分峰拟合,在532.1-532.5、530.6-530.8和529.4-529.7 eV均出现三个O 1s峰,分别归属为催化剂表面吸附的-OH或H2O中的氧(Oα2)、吸附在氧空位上的弱结合氧物种(Oα1)和晶格氧(Oβ)[34]。与Co3O4相比,Co3O4-RO的O 1s峰向低结合能处位移,结合XRD表征结果,这是由于Co3O4-RO晶胞参数变大,结晶度变差,导致O2-与Co离子的配位方式变化[35]。另外,表3给出了催化剂表面弱结合氧物种Oα1占整个氧物种数量的比值Oα1/(Oα1+ Oα2+ Oβ),显然,Co3O4-RO催化剂上Oα1所占的比例(0.34)明显高于Co3O4(0.23),表明还原-氧化预处理使Co3O4-RO表面上弱结合氧物种数量增加。
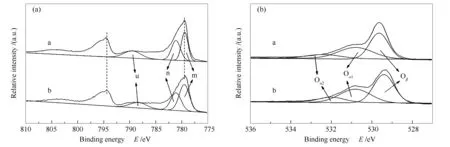
图 4 Co3O4和Co3O4-RO催化剂的XPS谱图

表 3 Co3O4和Co3O4-RO催化剂的XPS表征
2.5 催化剂氧气脱附能力表征
图5为预处理前后催化剂的O2-TPD谱图。
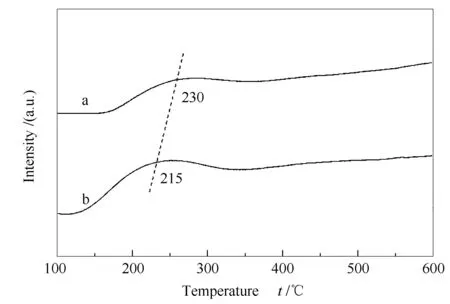
图 5 Co3O4和Co3O4-RO催化剂的O2-TPD谱图
由图5可知,两催化剂在150-300 ℃均出现一个脱附峰,可归属为表面氧物种的脱附。400 ℃以上的峰归属为晶格氧的脱附[36]。经还原-氧化预处理后,Co3O4-RO上氧气脱附起始温度和峰顶温度均明显向低温处位移,表明催化剂上的氧迁移率增加[37],表面氧物种更易于脱附,这是由预处理过程中Co3O4结构重建削弱Co-O键引起的[38]。一般来说,氧化物催化剂在通过氧化还原机理分解N2O时,表面氧的脱附被认为是催化N2O分解的速控步骤[36]。此外,根据两催化剂上O2脱附峰面积计算出Co3O4和Co3O4-RO的O2脱附量,其值分别为21.3和37.5 μmol/g。显然,Co3O4-RO的O2脱附量明显大于Co3O4,表明Co3O4-RO上可形成更多的氧空位。可见,对于尖晶石氧化物Co3O4而言,还原-氧化预处理不仅增强了其O2脱附能力,还可形成更多氧空位。
2.6 催化剂的反应性能评价
预处理前后Co3O4催化剂催化N2O直接分解活性结果见图6。由图6可知,Co3O4-RO具有相对较优异的催化活性,其可在300 ℃时实现 N2O完全转化,而未经预处理的Co3O4催化剂至少需要400 ℃。可见,还原-氧化预处理明显提高了Co3O4催化分解N2O活性。Co3O4上的N2O分子吸附、活化和分解等步骤可描述如下[12]:
N2O +* → N2O*
(5)
N2O* → N2+ O*
(6)
2O* → O2* → O2+*
(7)
N2O + O* → N2+ O2+ *
(8)
式中,*表示活性位。
首先,N2O分子吸附在活性位点上(式(5)),该过程中从催化剂表面给电子到N2O分子反键轨道,促使N-O键活化进而断裂解离为N2和吸附态的氧(式(6))。吸附的氧物种可通过L-H机理(式(7))或E-R机理(式(8))进行脱附。式(7)和(8)均为氧物种脱附、活性位再生步骤,其为整个N2O分解反应的速控步骤。对于L-H机理(式(7)),其核心是2O* → O2*,该过程与氧的流动性有关。根据O2-TPD的结果,与Co3O4相比,Co3O4-RO上氧气脱附起始温度和峰顶温度均明显向低温处位移,表明催化剂上的氧迁移率增加,即Co3O4-RO表面上的O*更容易结合为O2*进而完成脱附并实现活性位再生。
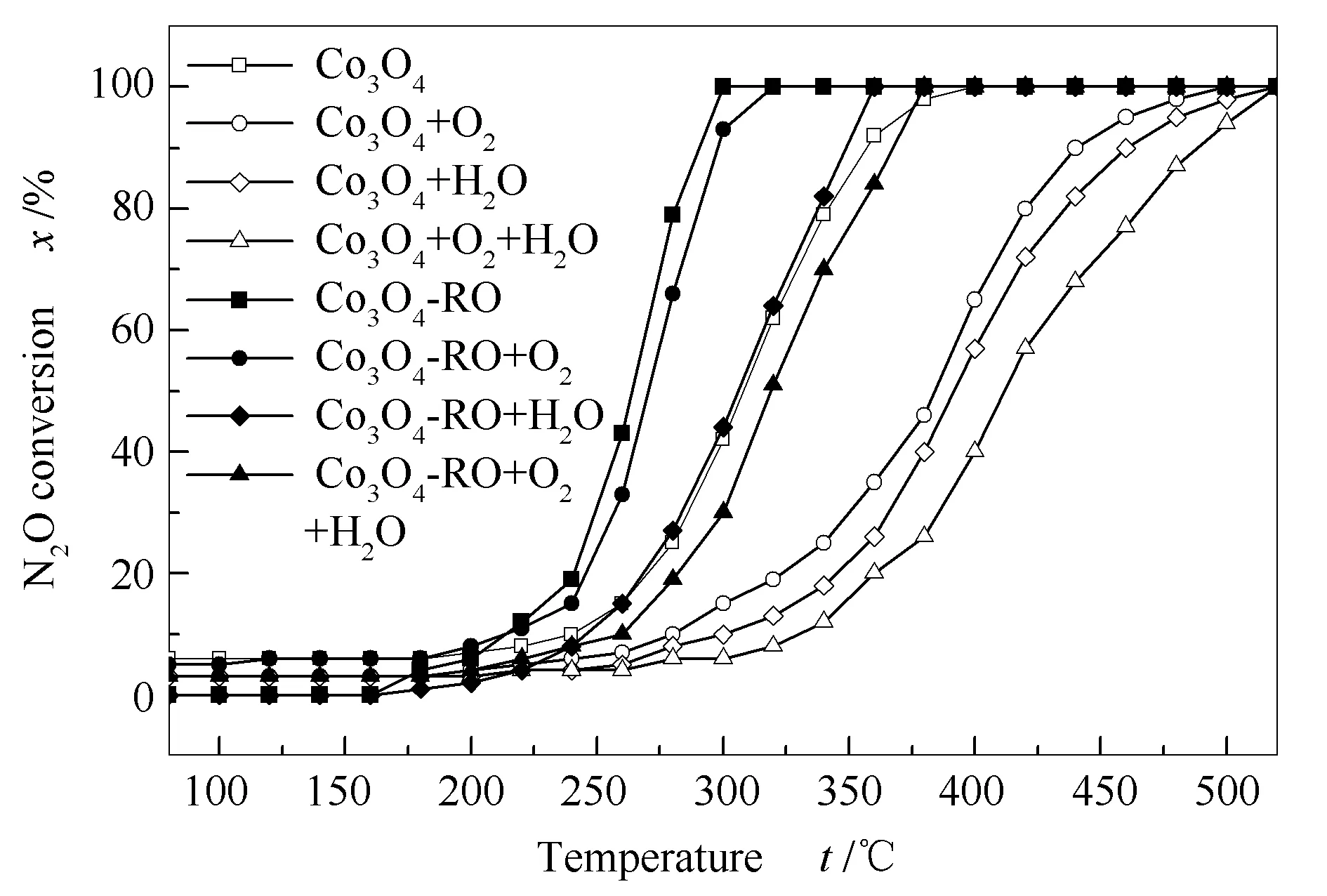
图 6 Co3O4和Co3O4-RO催化剂的活性评价
在实际生产中,原料气含有少量杂质气体,因此,催化剂对杂质气体良好的耐受性也尤为重要。图6还给出了原料气中杂质气体(O2和H2O)对催化剂催化活性的影响。由图6可以看出,O2对两种催化剂的催化活性均有抑制作用,这是由于O2的加入将使式(7)平衡向逆反应方向移动,阻碍两个O*的结合以及O2脱附和活性位点再生。但在O2存在条件下,Co3O4-RO的t100(360 ℃)仍明显低于Co3O4的t100(500 ℃)。根据O2-TPD表征结果,与Co3O4相比Co3O4-RO上的表面氧物种更易于脱附,因此,Co3O4-RO对O2表现出更强的耐受性。对于原料气中含有H2O的情况而言,Co3O4催化剂上N2O催化分解明显受到抑制,这是由H2O在活性位点上竞争吸附引起的。但在相同含H2O条件下,Co3O4-RO的催化活性仅略微下降。基于表2中O 1s XPS表征结果,Co3O4-RO上Oα2/(Oα1+Oα2+Oβ)值(0.10)低于Co3O4(0.13),表明Co3O4-RO表面上-OH和吸附水的浓度低于Co3O4。可见,还原-氧化预处理可在一定程度上抑制H2O在催化剂上的吸附,进而使Co3O4-RO对含H2O原料气有良好的耐受性。当原料气中加入2%O2+2.3%H2O时,Co3O4和Co3O4-RO催化剂的催化活性均有所抑制,但Co3O4-RO的催化活性仍明显高于Co3O4。图7给出了2%O2+2.3%H2O共存时两种催化剂的稳定性结果。由图7可知,Co3O4催化剂上N2O催化分解初始活性(约57%)仅维持5 h,此后N2O转化率逐渐下降,20 h后降低至39%。 然而,Co3O4-RO催化剂可在至少20 h内保持100%的N2O转化率。显然,在测试时间内,Co3O4-RO比Co3O4表现出更高的催化活性和稳定性。

图 7 Co3O4和Co3O4-RO催化剂的稳定性评价
2.7 动力学计算
金属氧化物催化剂上N2O分解为经典的一级反应[20]。表4为催化剂预处理前后的反应动力学数据。在260、280和300 ℃时,Co3O4-RO上N2O分解反应速率r明显高于相同反应温度下的Co3O4。例如,280 ℃时,Co3O4-RO上反应速率约为Co3O4的3.2倍(分别为0.090和0.028 μmol·s-1·g-1)。此外,如图8所示,根据阿伦尼乌斯公式计算出Co3O4-RO的活化能Ea(51.6 kJ/mol)低于Co3O4(75.9 kJ/mol)。可见经还原-氧化预处理后Co3O4-RO催化分解N2O反应的活化能明显降低。

表 4 Co3O4和Co3O4-RO的动力学参数

图 8 Co3O4和Co3O4-RO催化剂的lnk与1/T关系图
根据XRD和Raman表征结果,Co3O4催化剂经还原-氧化预处理结构重构后仍保持了尖晶石型结构,但结晶度变差,晶粒粒径减小,比表面积减小。H2-TPR表征发现,Co3O4-RO催化剂氧化还原能力增强,说明还原-氧化预处理在使催化剂结构重建的同时,削弱了Co-O键,这有利于促进表面氧空位的形成和O2的脱附。XPS结果证实了经还原-氧化预处理后,催化剂表面弱结合氧物种数量增加。O2-TPD结果进一步表明,经还原-氧化预处理的催化剂具有更强的O2脱附能力和更多的O2脱附量。因此,还原-氧化预处理不仅导致催化剂结晶度变差,晶粒粒径减小,同时因结构重建削弱了Co-O键,弱结合氧物种数量增多,更重要的是使催化剂表面的氧物种更易于脱附,降低了反应活化能,因而Co3O4-RO具有优异的催化N2O分解性能。
3 结 论
研究了还原-氧化预处理对Co3O4催化剂晶相结构、织构、氧化还原能力和表面性质等的影响,同时考察了催化剂催化分解N2O性能。还原-氧化预处理过程中催化剂发生结构重构,不仅导致催化剂结晶度降低,晶粒粒径减小,尤其是削弱了Co-O键,增强了催化剂表面的氧脱附能力,使得其催化分解N2O性能提高。因此,还原-氧化预处理氧化物催化剂,对于表面氧脱附为速控步骤的反应具有积极促进作用,同时还对原料气中O2和H2O表现出良好的耐受性。
猜你喜欢
材料与冶金学报(2022年2期)2022-08-10
温州大学学报(自然科学版)(2022年2期)2022-05-30
金属热处理(2022年2期)2022-03-16
安徽工业大学学报(自然科学版)(2021年3期)2021-09-08
粉末冶金技术(2021年3期)2021-07-28
铝加工(2021年2期)2021-05-17
建材发展导向(2021年23期)2021-03-08
有色金属科学与工程(2021年1期)2021-03-04
装备维修技术(2020年5期)2020-11-20
分析化学(2018年1期)2018-01-18
