
MOFs衍生炭负载的钴基催化剂的廉价制备及其CO加氢催化性能
2019-04-18 08:57马彩萍张成华李永旺
燃料化学学报 2019年4期
李 宁, 马彩萍, 张成华, 杨 勇, 李永旺
(1. 中国科学院山西煤炭化学研究所 煤转化国家重点实验室, 山西 太原 030001; 2. 中国科学院大学, 北京 100049; 3. 中科合成油技术有限公司 国家能源煤基液体燃料研发中心, 北京 101407)
中国能源结构表现出“富煤、少气、缺油”的显著特点,石油消费大量依赖进口,过去五年间的年均石油进口量占消费总量的64.87%[1]。费托合成反应可将丰富的煤炭资源经合成气转化为清洁的液体燃料[2-4],进而缓解中国石油短缺的压力。费托反应催化剂主要包括钴基催化剂和铁基催化剂,前者具有更高的费托反应活性、较高的C5+产物选择性、较低的水煤气变换活性,被广泛应用于工业生产[5-7]。但是金属Co储量较少、价格昂贵,工业上通常将其与载体复合组成负载型催化剂以降低成本,载体的性质会显著影响其催化活性及稳定性。
诸多研究采用氧化物作载体,包括Al2O3、SiO2和TiO2等,但该类载体与金属活性组分间相互作用强,易生成难还原的复合氧化物,进而导致催化剂发生不可逆失活[8-11]。采用活性炭、碳纳米管及碳纳米纤维等惰性炭材料作载体能够避免复合氧化物的生成,但其与金属间的相互作用较弱,在反应后期金属纳米颗粒容易聚集烧结[11-13]。近年来,金属有机框架材料(Metal-Organic Frameworks,MOFs)衍生制备核壳结构(Co@C)的炭载催化剂受到广泛关注,其良好的限域作用能够有效抑制催化剂烧结失活[14-20]。
MOFs是由无机金属离子与有机配体通过氢键与配位键自组装而成的周期性网络骨架结构[21],对苯二甲酸(H2BDC)价廉易得,是最早被应用于合成MOFs材料的有机配体[22],但是由于其水溶性差,通常需要使用昂贵且毒性大的有机溶剂溶解,增加了MOFs合成及后处理的成本。由于H2BDC易溶于碱性水溶液,使得在廉价环保的水溶剂中来合成BDC MOFs材料成为可能,这要求有准确的H2BDC在水溶液中溶解度数据,但目前相关报道较少。本研究首先测定了H2BDC在KOH、NaOH等碱性水溶液中的溶解度数据,然后采用共沉淀法大量合成Co-BDC MOFs,以其为前驱体通过化学气相沉积法制备Co@C催化剂,考察炭化气氛(Ar和C2H2)对催化剂结构和组成的影响,并对其进行费托反应性能评价。
1 实验部分
1.1 Co-BDC MOFs的制备
分别配制浓度为0.2-5.0 mol/L的NaOH、KOH、NaHCO3、Na2CO3、NH3·H2O标准溶液,加入适量H2BDC,于25 ℃恒温搅拌20 min,每个数据点取三次测量平均值,记录溶解度数据。参照上述步骤,分别记录40和55 ℃时,H2BDC在KOH标准溶液中的溶解度数据。
取适量H2BDC溶于KOH溶液、C4H6CoO4·4H2O溶于水中,并采用CH3COOH溶液调节其酸碱度,在一定温度下共沉淀,相同温度下老化24 h后过滤洗涤,将所得产物于120 ℃下干燥12 h,压片、破碎、筛分得到催化剂前驱体(20-40目)。
1.2 Co@C催化剂的制备
取适量前驱体分别在Ar和2%C2H2/Ar气氛中以5 ℃/min的速率升温至550 ℃炭化1 h,降至室温后用无水乙醇保护,所得催化剂分别记为Co@C-Ar、Co@C-C2H2。
1.3 Co@C催化剂的表征
材料物相结构表征在德国Bruker公司的 AX-D8 X射线粉末衍射仪上进行。 X射线源采用 CoKα(γ=0.178 nm),管电压为35 kV,管电流40 mA,步长0.04°。
样品的织构性质采用ASAP 2420物理吸附仪(Micromeritics,美国)进行测定。样品在测试前均进行真空脱气。其中,Co-BDC MOFs材料均在70 ℃下真空脱气12 h,催化剂均在160 ℃下真空脱气2 h, 然后在350 ℃下真空脱气8 h。比表面积、孔容以及孔道结构分别采用Brunauer-Emmett-Teller(BET)、t-plot和Berrett-Joyner-Halenda (BJH)法计算。
扫描电子显微镜(SEM)照片采用Quanta 400场发射扫描电子显微镜(FEI,美国)拍摄,测试电压为30 kV。
透射电子显微镜(TEM)照片采用TalosTM 200 A型电子显微镜(FEI,美国)拍摄,加速电压为200 kV。
样品表面元素采用美国Thermo Escalab 250Xi型X射线光电子能谱仪(AlKα射线,多通道检测仪)进行分析。测试结果经C 1s结合能(284.8 eV)进行校准后以高斯/洛伦兹(70/30)进行分峰拟合后分析。
TG分析采用METTLER TOLEDO公司的TGA/DSC 1型热重分析仪。取20 mg样品置于Al2O3坩埚中,分别在C2H2和Ar气氛下以10 ℃/min的速率升温至900 ℃,记录质量失重百分率与温度间的关系。
拉曼光谱(Raman)分析采用LabRAM-800型拉曼光谱仪,Ar-离子激光器波长为514 nm,强度为5 mW,800-2000 cm-1扫描。
1.4 Co@C催化剂的评价
催化剂的费托合成反应在固定床反应器中进行。取20-40目催化剂颗粒装入反应管恒温区。反应前所有催化剂在H2氛围中于240 ℃ 还原24 h(空速2000 h-1、压力0.5 MPa)。还原后降温至150 ℃,以H2/CO体积比为2.0的合成气为原料提压后升温至220 ℃,在压力3.0 MPa,空速3000 h-1的反应条件下进行F-T活性及其反应运行稳定性测试。反应后催化剂在乙醇保护下由反应管中取出进行表征。固相和液相产物分别经热阱(160 ℃)和冷阱(0 ℃)收集,其中,气相产物采用气相色谱仪(Agilent 6890N)进行在线分析。
2 结果与讨论
2.1 Co-BDC MOFs的制备
首先研究了碱溶液种类及浓度对H2BDC溶解度的影响。H2BDC在NaOH、KOH、NaHCO3、Na2CO3、NH3·H2O溶液中的溶解度曲线见图1。由图1可知,H2BDC的溶解度随NaHCO3溶液浓度增大而增大,在其余四种碱溶液中溶解度与碱溶液浓度曲线均呈 “火山型”,但在不同碱溶液中溶解度随浓度变化速率不同,说明H2BDC溶解度受碱溶液的种类及浓度的影响较为显著。其中,在3 mol/L的KOH溶液中其溶解度最大,为18 g H2BDC/100 g KOH溶液。以KOH溶液为例测定了H2BDC溶解度随温度的变化,见图2。由图2可知,H2BDC溶解度随温度升高而小幅增大,说明温度对H2BDC溶解度影响较小。综合考虑,以3 mol/L的KOH溶液为溶剂在室温下溶解H2BDC。
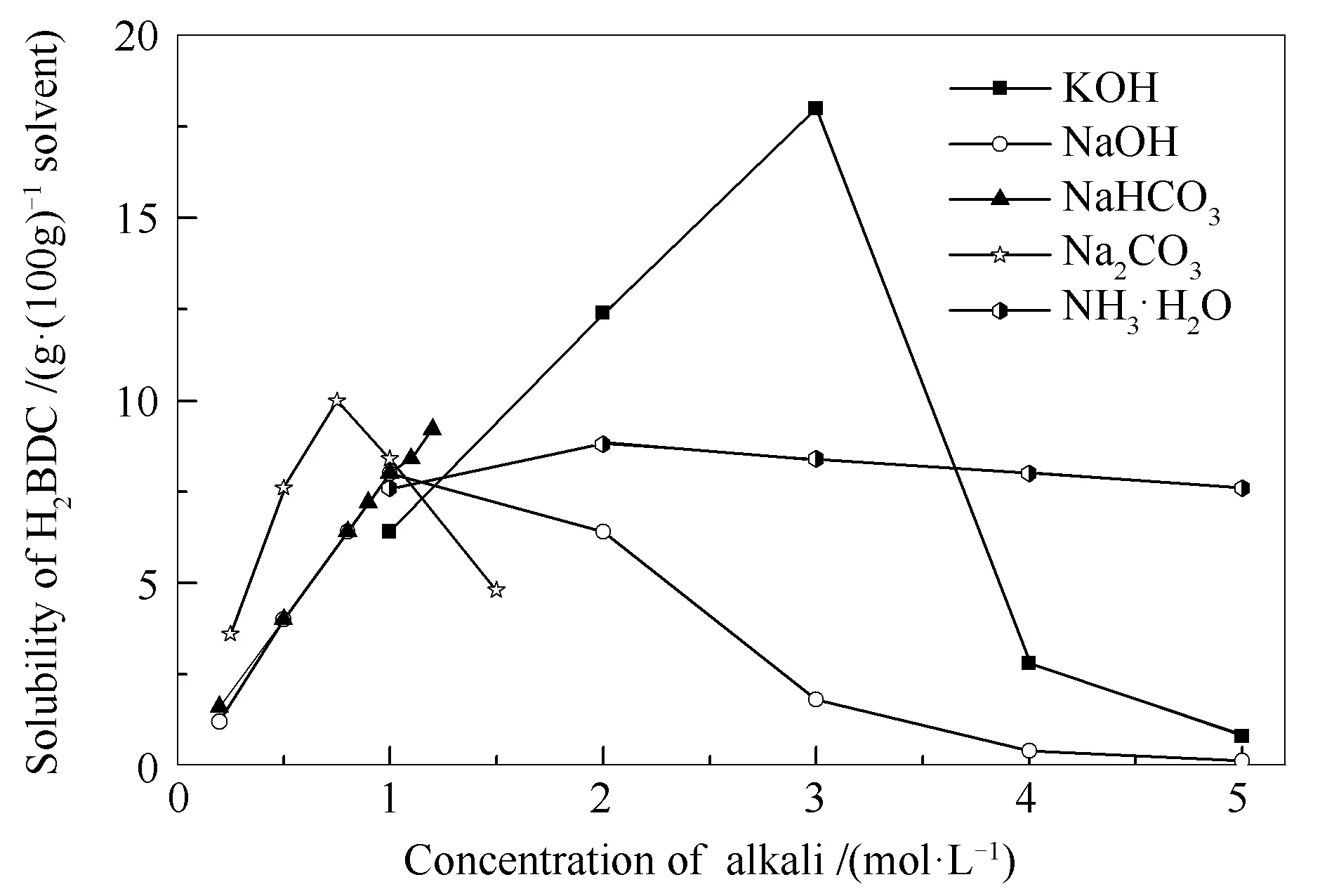
图 1 H2BDC在碱溶液中溶解度(25 ℃)
共沉淀反应中溶液的pH值、反应物配比以及反应温度是影响样品结晶度的三个重要因素。不同pH值、反应物配比和不同温度下合成样品的XRD谱图见图3。由图3(a)可知,在碱性条件下(pH=8.0-9.0)合成样品在10.6°、18.1°、23.2°、32.1°、34.6°、42.2°、45.4°、52.6°、59.5°、70.3°、77.7°处出现归属于Co+3O(OH)沉淀特征衍射峰(PDF#07-0169);在弱酸性条件下(pH=5.0-6.0)和中性条件下(pH=7.0)样品的XRD谱图一致,在10.8°和20.7°处出现特征衍射峰,归属于Co-BDC MOFs[23,24],表明中性及弱酸性条件有利于Co-BDC MOFs生成。然而当调节反应液的pH=4时,则会生成Co-BDC MOFs与大量对苯二甲酸晶体的混合产物,表明pH值小于5的反应条件不利于MOFs晶体生成,不仅会导致MOFs产量降低而且增加了MOFs后处理难度。这是因为调节pH值至2-4时,溶解于碱溶液中的对苯二甲酸会大量析出,结晶析出的对苯二甲酸难以与Co2+配位形成MOFs材料[25,26]。为了改善材料的结晶度、增大比表面积,在弱酸性条件下研究反应物配比(物质的量比H2BDC∶Co=3∶1,2∶1,1∶1,1∶2,1∶3)和反应温度(70、80、90 ℃)对材料结构的影响。由图3(b)可知,随物质的量比减小Co-BDC MOFs的特征衍射峰强度增强,说明Co前驱体过量时所得Co-BDC MOFs骨架结构结晶度较高,这是由于反应体系中过量的金属离子有利于晶核长大。由图3(c)可知,Co-BDC MOFs的特征衍射峰强度随反应温度升高而增强,表明升高温度有利于提高Co-BDC MOFs材料结晶度。样品的织构性质见表1,由表1可知,Co-BDC MOFs比表面积与孔容积相对较大的合成条件为pH=5、80 ℃、H2BDC∶Co=1∶1(物质的量比),但考虑到配体过量的反应条件能够为后期炭化提供足够的碳源,选择在H2BDC∶Co=2∶1(物质的量比)条件下进行MOFs合成,其他反应条件选择pH=5、80 ℃,以保持其较大的比表面积。
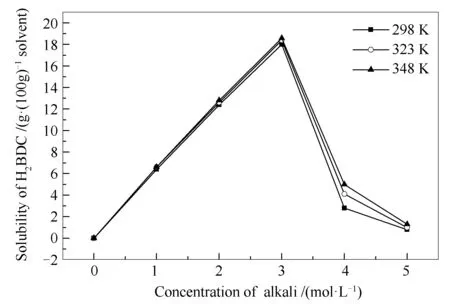
图 2 不同温度下H2BDC在KOH溶液中的溶解度
Co-BDC MOFs前驱体的表征结果见图4。由图4(a)可知,此条件下合成的样品在2θ=10.8°和20.7°处出现归属于Co-BDC MOFs的特征衍射峰;由图4(b)可知,吸附-脱附曲线为典型的IV型吸附等温线,存在较大的回滞环,说明样品中存在大量介孔,比表面积为62 m2/g;由图4(c)和(d)的SEM照片可以看出,此材料为薄片状结构,表明在廉价环保的水溶剂中可以制备出包含较多介孔的薄片状Co-BDC MOFs材料。水作溶剂不仅绿色环保,还能够降低MOFs合成及后处理成本,为BDC MOFs合成开发提供了新的研究思路。
a: other synthesis condition: 70 ℃, H2BDC∶Co=2∶1(molar ratio);
b: other synthesis condition: pH=6, H2BDC∶Co=2∶1(molar ratio);
c: other synthesis condition: 70 ℃, pH=6
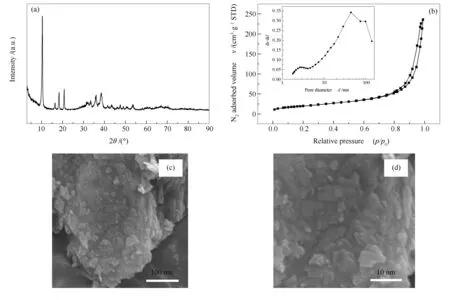
图 4 Co-BDC MOFs的XRD谱图(a)、N2吸附-脱附等温曲线(b)和扫描电镜照片((c), (d))
2.2 Co@C催化剂的制备
以Co-BDC MOFs前驱体为牺牲模板在C2H2和Ar气氛下一步炭化制备Co@C催化剂。TG曲线用来研究Co-BDC MOFs质量随温度的变化情况,具体见图5。
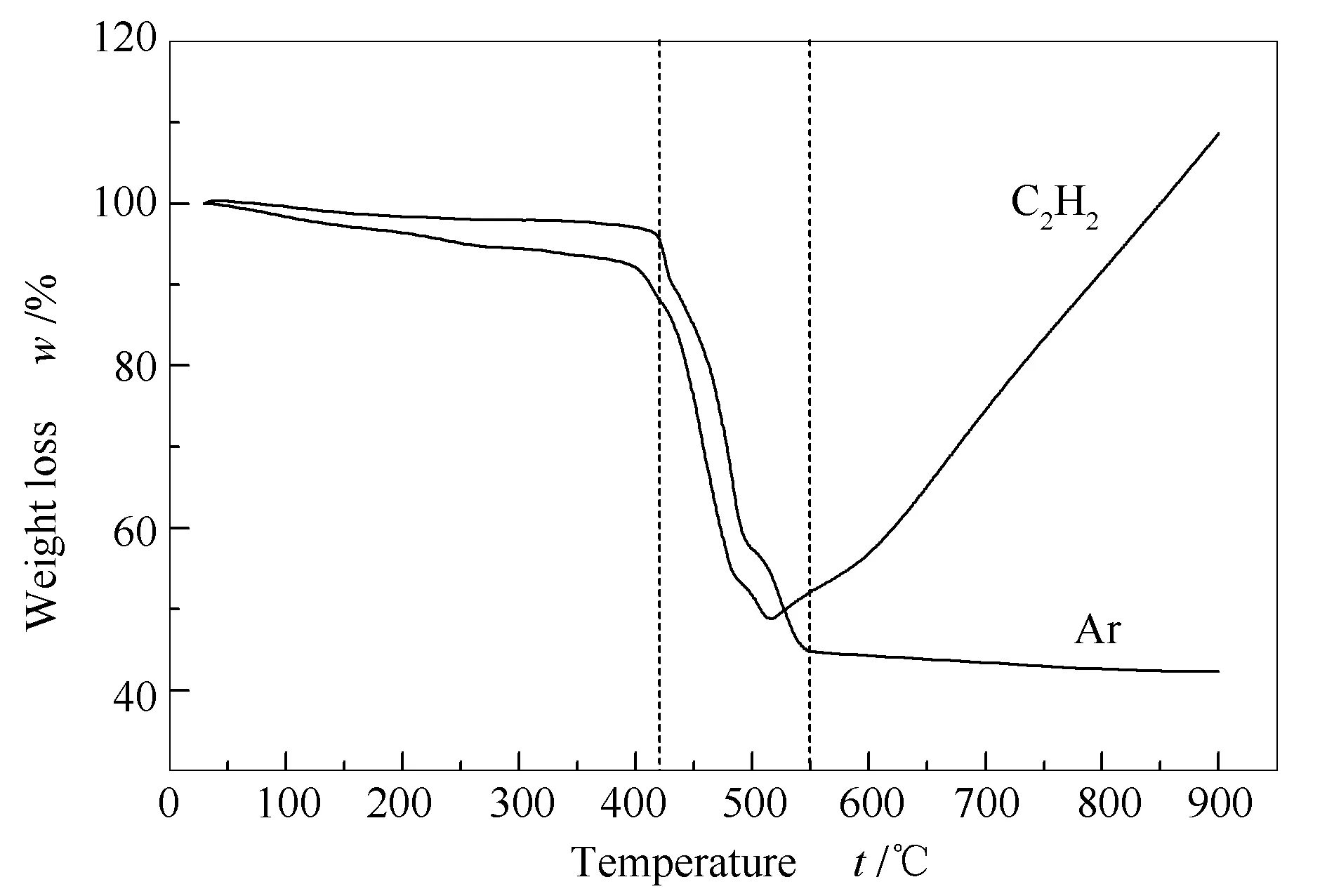
图 5 Co-BDC MOFs在C2H2 and Ar气氛下的热重曲线
由图5可知,Ar气氛下样品质量变化分为三个阶段:第一阶段(150-410 ℃)失重率约为8.98%,主要为样品中参与配位水的失重;第二阶段(410-550 ℃)失重率约为43.24%,主要为MOFs骨架中有机配体分解的失重;第三阶段为少量残余配体继续分解的失重。C2H2气氛下样品前两阶段的质量变化与前述Ar气氛一致,不同的是,第三阶段C2H2气氛下样品出现了积炭增重现象,说明C2H2可能作为碳源参与炭化过程。失重数据表明在550 ℃时 MOFs骨架基本坍塌。因此,在550 ℃,Ar和2%C2H2/Ar气氛下对样品Co-BDC进行1 h炭化处理,得到Co@C-Ar、Co@C-C2H2催化剂。
2.3 Co@C催化剂的表征
催化剂的TEM照片见图6。由图6(a)、(c)可知,两种催化剂粒子分布均匀;由HRTEM(图6(b)、(d))照片中均可看到明显的晶格条纹,晶格间距0.20 nm对应于金属Co(fcc)核,晶格间距0.34 nm对应于石墨化的炭壳结构,表明炭化后形成Co@C核壳结构。催化剂Co@C-Ar的炭壳层较厚,这是因为在Ar气氛下炭化时Co离子周围的桥连配体不断热解转化为多层致密的石墨炭壳包裹在Co纳米颗粒上[27];而催化剂Co@C-C2H2的炭壳层较薄,因为C2H2气氛中炭化时样品表面碳元素不断吸附沉积同时与分解的有机配体相互作用,从而形成低石墨化程度的薄层石墨炭壳[20]。催化剂粒径统计分布见图7。由图7可知,Co@C-Ar和Co@C-C2H2的平均晶粒粒径分别为15.6和13.6 nm,其尺寸相差不大。
催化剂的元素含量及织构性质信息见表2。由表2可知,两种催化剂的表面含碳量明显高于前驱体MOFs,表明在炭化过程中MOFs有机骨架坍塌形成炭载体。Co@C-C2H2和Co@C-Ar催化剂表面含碳量分别为83.15%和75.78%,前者的表面碳含量更高,说明C2H2气氛中的碳元素在样品表面沉积参与样品炭化过程。Co@C-C2H2和Co@C-Ar的比表面积分别为137和86 m2/g,孔径分别为13.3和11.0 nm,容积分别为0.25和0.23 cm3/g,前者具有更大的比表面积、孔径及孔容积,表明C2H2气氛不仅能够为MOFs炭化提供外加碳源,还有利于增大催化剂的比表面积。

表 2 Co-BDC MOFs和Co@C-Ar、Co@C-C2H2催化剂的性质
a: analysed by XPS
催化剂的XRD谱图见图8。由图8可知,两种催化剂均在51.8°、60.6°和48.7°、55.7°处出现特征衍射峰,分别归属于fcc Co物相(PDF#89-4307)和hcp Co物相(PDF#89-7373),表明Co-BDC MOFs中Co离子在炭化过程中能够被还原为金属态且炭化气氛对Co物相结构的形成影响不大。
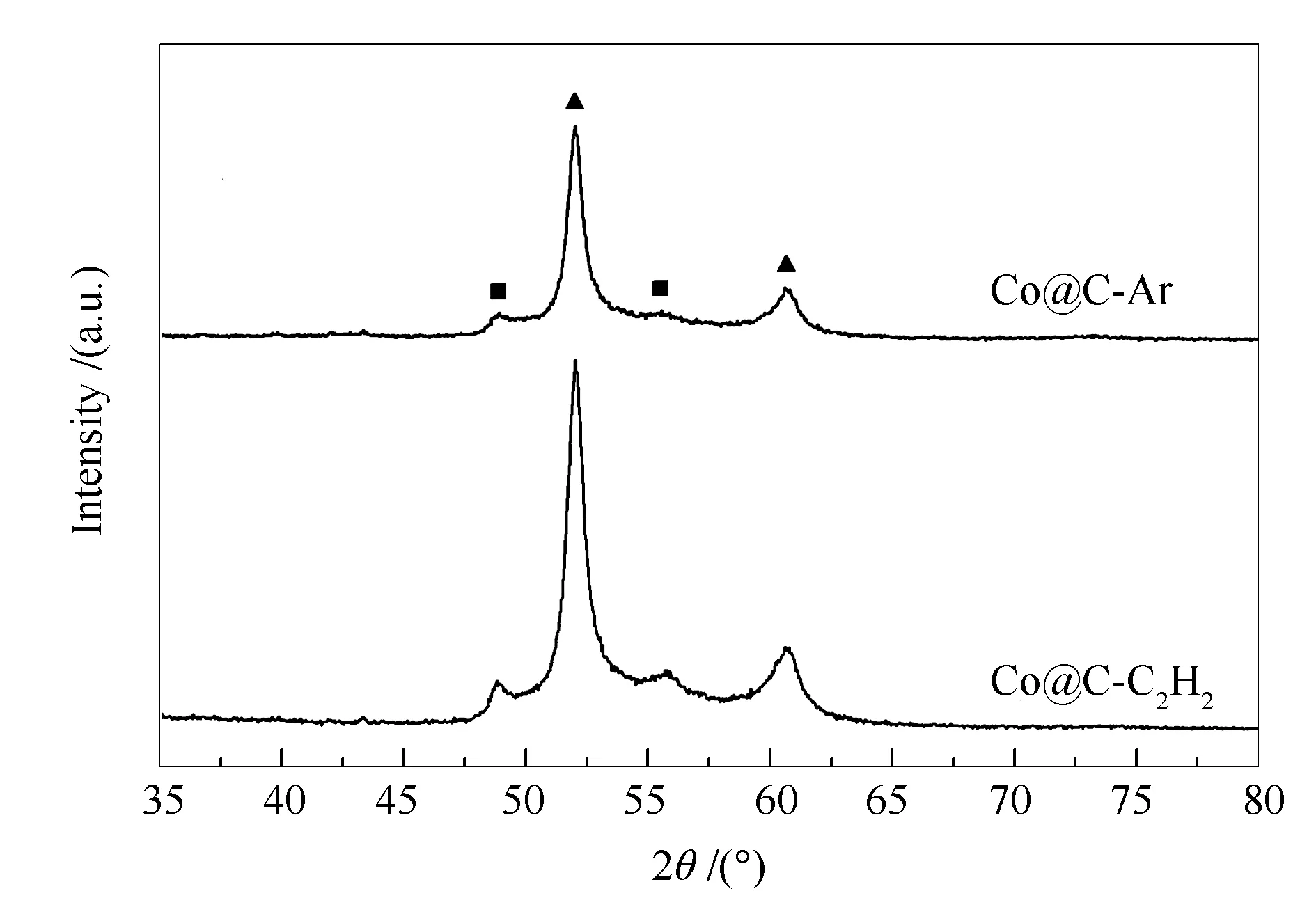
图 8 催化剂Co@C-C2H2和Co@C-Ar的XRD谱图
拉曼光谱可用来表征炭层结构的石墨化程度,催化剂的拉曼光谱谱图见图9。由图9可知,1347 cm-1处的D峰归属于存在晶格缺陷的无序炭结构,1590 cm-1处的G峰归属于石墨化的有序炭结构,D峰与G峰的强度之比(ID/IG)能够表征炭材料的石墨化程度,ID/IG越高炭层结构的石墨化程度越低。催化剂Co@C-Ar和Co@C-C2H2的ID/IG值分别为0.93和1.00,表明Co@C-C2H2样品的炭层结构较为疏松,石墨化程度更低,与TEM表征结果一致。研究表明,疏松多孔的炭壳有利于为反应物及产物进出提供更多孔道[20],从而提高费托反应活性以及促进长链烃产物生成。

图 9 催化剂Co@C-Ar和Co@C-C2H2的拉曼光谱谱图
2.4 催化剂的性能评价
催化剂的费托反应性能结果见图10。由图10可知,两种催化剂在反应过程中活性稳定,表明Co@C核壳结构有利于提高炭载钴基催化剂的稳定性。Co@C-C2H2催化剂的CO转化率、C5+长链烃选择性以及CH4选择性分别为10.50%、82.66%、15.29%;Co@C-Ar催化剂的CO转化率、C5+产品选择性以及CH4选择性分别为6.22%、66.49%、22.28%。前者表现出更高的CO转化率和C5+长链烃选择性,这是由于在C2H2气氛下炭化时形成的石墨炭层结构较为疏松多孔,有利于长链烃产物的生成[20]。
反应后催化剂的TEM照片和粒径分布分别见图11和图12。由图11和图12可知,两种催化剂粒子分散均匀且无明显聚集烧结,Co@C-Ar和Co@C-C2H2催化剂中Co纳米颗粒的平均晶粒粒径分别为16.9和14.6 nm,与反应前相比并未发生明显改变,表明炭壳层有明显的限域作用,因此,催化剂在反应过程中稳定性较好。HRTEM照片中出现明显的晶格条纹,晶格间距0.22 nm,对应Co2C的(110)晶面,表明催化剂体相存在Co2C。
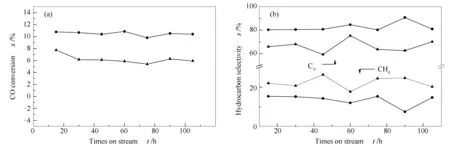
图 10 催化剂Co@C-Ar和Co@C-C2H2的CO转化率(a)及烃选择性(b)的变化

图 11 催化剂Co@C-C2H2((a), (b))和 Co@C-Ar((c), (d))反应后的TEM照片
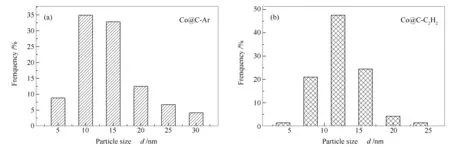
图 12 反应后的催化剂Co@C-Ar(a)和Co@C-C2H2(b)的粒径分布
为进一步探究催化剂的物相转变,将反应后的催化剂进行XRD表征,见图13。
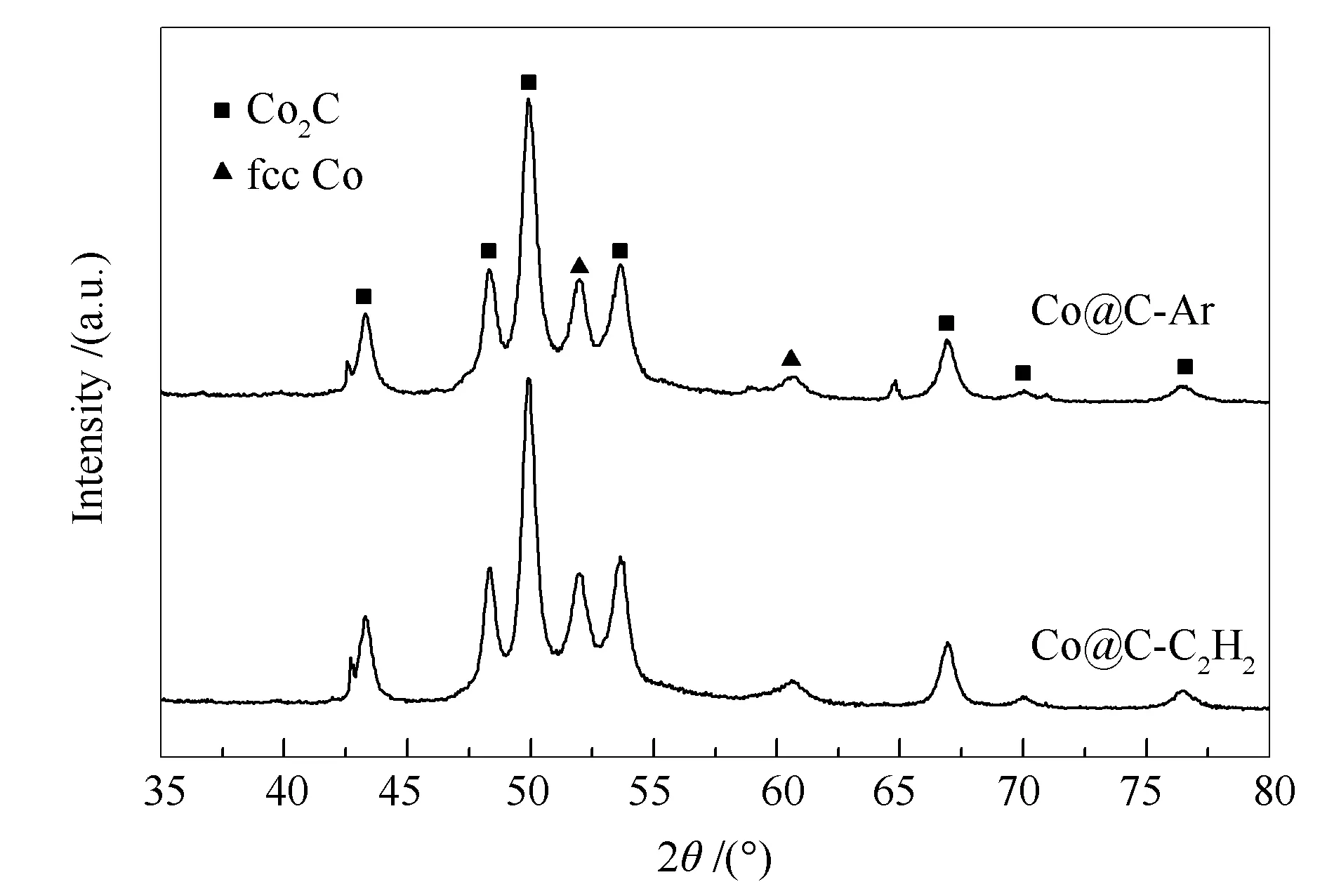
图 13 Co@C-C2H2和 Co@C-Ar在费托反应后的XRD谱图
与反应前的催化剂相比,在53°和60°处的fcc Co物相(PDF#89-4307)的特征衍射峰强度减弱,hcp Co物相的特征衍射峰消失,在43.2°、48.3°、49.9°、53.7°、66.3°、70.1°、76.3°处出现归属于Co2C物相的特征衍射峰(PDF#72-1369),表明催化剂中全部的hcp Co物相和部分的fcc Co物相转化为Co2C。催化剂由最初的活性相金属Co逐渐转变为无特殊形貌的Co2C后仍然保持稳定的反应活性,可以推断Co2C也具有费托反应活性,但这与文献报道并不一致。许多研究表明,Co2C不具备费托反应活性,其生成会导致催化剂的失活[28,29];同时,也有研究表明暴露特定晶面Co2C物相具有优异的费托反应活性和选择性[30,31],但本实验的结果表明,催化剂的反应前后的物相由金属钴物相转变为Co2C物相,其费托合成反应活性并未发生明显变化,且这些Co2C颗粒并未显示出特定的晶面暴露现象,说明普通的Co2C纳米粒子与金属钴一样,同样具有相当的费托合成活性。
3 结 论
在碱性水溶液中通过共沉淀法大量合成Co-BDC MOFs前驱体,以其为牺牲模板通过化学气相沉积法分别在C2H2和Ar气氛制备Co@C核壳结构催化剂。XRD结果表明,炭化气氛对金属Co物相形成及粒子粒径影响较小;TEM和Raman表征结果表明,炭化气氛对炭壳结构的石墨化程度影响较大,C2H2气氛有助于形成疏松多孔的石墨炭层结构从而有利于长链烃产物的生成,Co@C-C2H2催化剂的C5+烃选择性高达80%。反应前后催化剂的TEM结果表明,两种催化剂颗粒粒径分布均匀且反应后催化剂未发生明显的烧结,说明炭壳结构限域作用显著。催化剂由反应前的单相金属Co转化为金属Co与Co2C的混合相后未发生失活,表明Co2C也具有费托反应活性。
致谢
本项目感谢中科合成油术有限公司在设备和资金方面给予本研究工作的鼎力支持。
猜你喜欢
林产工业(2022年2期)2022-03-05
农业研究与应用(2021年2期)2021-08-12
世界有色金属(2020年4期)2020-05-16
东南文化(2020年1期)2020-04-27
燃料化学学报(2019年10期)2019-11-04
科技与创新(2018年10期)2018-05-23
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
中学生数理化·中考版(2015年11期)2015-09-10
中学化学(2015年5期)2015-07-13
