
Crim1过表达抑制肥大乳鼠心室肌细胞瞬时外向钾电流改变
2019-03-30 01:29
国际心血管病杂志 2019年6期
病理性心肌肥大导致的心室重构是慢性充血性心力衰竭(CHF)最主要的病理生理机制[1-2]。心脏性猝死是CHF患者主要的死亡原因[3]。心室肌细胞离子通道重构是导致心室肌细胞动作电位改变,进而引发恶性室性心律失常的重要病理生理基础[4-5]。
半胱氨酸丰富跨膜成骨蛋白调控因子(Crim1)是调控胚胎组织器官发育的重要因子,并参与心室肌细胞及心室组织肥大的调控[6-7]。Crim1是否参与肥大心肌细胞离子通道重构的调控目前尚不清楚。研究发现,在培养的肥大乳鼠心室肌细胞模型中,编码瞬时外向钾电流(Ito)离子通道α亚基的Kv4.2基因和蛋白表达下调,心室肌细胞Ito离子通道失活加快,而致Ito电流密度减小,细胞动作电位时程(APD)延长[8]。本研究旨在探讨Crim1过表达对肥大心室肌细胞Ito的调控作用。
1 对象与方法
1.1 主要试剂和仪器
兔抗鼠Crim1抗体(北京博奥森公司),兔抗鼠甘油醛-3-磷酸脱氢酶(GAPDH)抗体(Santa Cruz公司),羊抗兔辣根过氧化物酶-IgG(北京中杉金桥公司),胰酶和Ⅱ型胶原酶(Sigma公司)。高糖DMEM培养基、特优级胎牛血清(FBS)、苯肾上腺素(PE)、5-溴脱氧尿嘧啶核苷(5-BrdU)均为Gibco公司产品。携带Crim1基因的重组腺病毒(Ad-Crim1)、腺病毒空载体(Ad-null)购自上海ThermoFisher SCIENTIFIC公司。Axopatch 700B膜片钳放大器及Digidata 1322数据转换器(美国Axon公司),Sutter p-97微电极拉制仪(美国Sutter公司),BJ-40玻璃微电极(北京正天易科贸有限公司)。
记录钾电流的细胞外液(mmol/L):NaCl 136,HEPES 10.0,KCl 5.4,MgCl2·6H2O 1.0,Glucose·H2O 10.0,NaH2PO40.33,BaCl20.5,CdCl20.3,CaCl2·2H2O 2.0,用NaOH溶液将pH值调至7.4。电极内液(mmol/L):Na2·ATP 5.0,KCl 140,HEPES 10.0,MgCl2·6H2O 1.0, EGTA 5.0,用KOH溶液将pH值调至7.2。含钙台氏液(mmol/L):NaH2PO40.33,NaCl 136,KCl 5.4,葡萄糖 10.0,HEPES 10.0,CaCl2·2H2O 2.0,用NaOH溶液将pH值调至7.4。
1.2 实验动物
清洁级1 d龄SD大鼠乳鼠,雌雄不限,购于北京大学医学部动物中心[许可证号SYXK(京)2011-0039]。此动物实验通过贵州省人民医院伦理委员会批准(院伦理审查[2012]001号)。
1.3 原代乳鼠心室肌细胞的分离和培养
乙醚气雾麻醉下处死乳鼠,75%乙醇体表消毒。分离左心室,保留室间隔。剪碎心肌组织,给予终浓度分别为0.1%的胰蛋白酶和0.03%的2型胶原酶混合酶解液分离细胞。差速贴壁与BrdU结合处理获得纯化的心肌细胞[7,9-10]。加入10%FBS-DMEM培养基,于37 ℃ 5% CO2培养箱中培养48 h后换成无血清DMEM高糖培养基进行后续实验。
1.4 重组腺病毒感染原代心室肌细胞的有效性鉴定
细胞培养48 h后换成无血清DMEM高糖培养基,分4组,分别给予Ad-null和Ad-Crim1 [分3组,感染复数(MOI)分别为25、100、200] 感染原代心室肌细胞32 h,Western blot检测Crim1蛋白表达,验证Ad-Crim1感染过表达的有效性,并据此筛选出合适的病毒感染滴度用于后续实验。
1.5 细胞分组干预
心室肌细胞培养48 h后更换无血清DMEM高糖培养基,按干预方式不同分为3组。重组腺病毒空载体(Ad-null)组:Ad-null感染细胞32 h。重组腺病毒空载体+苯肾上腺素(Ad-null+PE)组:Ad-null感染细胞8h后,予PE干预24 h。Crim1过表达的重组腺病毒+苯肾上腺素(Ad-Crim1+PE)组:Ad-Crim1感染细胞8 h后,予PE干预24 h。
1.6 心室肌细胞结晶紫染色
结晶紫行活细胞染色。每组细胞在400倍显微镜下随机拍照10个视野,ImageJ软件测量细胞表面积,计算细胞平均面积。
1.7 Western blot检测
提取总蛋白。细胞蛋白按40 μg上样量进行电泳。转膜后用5 %羊血清37 ℃封闭2 h,加入一抗兔抗鼠Crim1抗体(1∶100)或兔抗GAPDH(1∶1 000),4 ℃孵育过夜,再与二抗羊抗兔辣根过氧化物酶-IgG,37 ℃孵育2 h。Bio-Rad化学发光仪进行检测。利用ImageJ专业图像分析软件对发光条带进行半定量分析,以GAPDH为内参进行校正。每组样本重复3次实验。
1.8 全细胞膜片钳检测心室肌细胞Ito
预温的0.125%胰酶消化心室肌细胞,制成单细胞悬液。于直径35 mm培养皿中调整细胞数约为1×102个/mL,37℃ 95% CO2培养2~3 h使细胞贴壁。选择立体感强,大小适中的细胞进行实验。玻璃微电极充灌电极液后电阻为2~4 MΩ。施加负压使电极与细胞表面形成1 GΩ以上高阻抗封接。破膜,给予慢电容补偿,形成全细胞记录。设置钳制电压为-80 mV,给予指令电位从-40~+70 mV,跃阶电压10 mV,波宽300 ms,频率0.2 Hz的刺激,记录Ito。5 mmol/L的4-氨基吡啶能阻断该电流,证实该电流为Ito。为避免因细胞大小所造成的误差,采用电流密度分析,电流密度(pA/pF)=电流强度/电容。电流信号经Ag/AgCl电极引导,由膜片钳AXON 700B放大器放大,通过AD/DA转换板,存储于计算机硬盘中。实验过程由pCLAMP 10.0软件程进行序刺激发放和信号采集。
1.9 统计学处理
采用pCLAMP 10.0软件进行数据和图形转换,运用SigmaPlot软件绘制离子通道电流密度-电压曲线。采用SPSS 19.0软件进行统计分析。数据以均数±标准差表示。应用单因素方差分析进行多组间比较,多组间两两比较方差齐性时用LSD检验,方差不齐性时用Dunnett T3检验,P<0.05为差异有统计学意义。
2 结果
2.1 Ad-Crim1感染心室肌细胞促Crim1蛋白表达
为证实Ad-Crim1感染原代心室肌细胞促Crim1蛋白表达的有效性,分别用Ad-Crim1或者Ad-null感染原代心室肌细胞32 h。当Ad-Crim1滴度为分别MOI 25、100及200时,Ad-Crim1 3组心室肌细胞Crim1蛋白相对表达水平均较Ad-null组(Ad-null组为1)明显升高,分别为1.83±0.13、2.51±0.12和2.82±0.27(P均<0.05),说明Ad-Crim1感染原代心室肌细胞促Crim1蛋白表达有效。Ad-Crim1 3组中MOI 25组Crim1蛋白表达水平较MOI 100组和MOI 200组低,而MOI 100组和MOI 200组表达水平相近。因此,后续实验选取Ad-Crim1感染心室肌细胞的病毒滴度为MOI=100。见图1。

图1 腺病毒感染培养的心室肌细胞促Crim1蛋白表达
2.2 过表达Crim1抑制PE诱导的心室肌细胞肥大
结晶紫染色显示Ad-null组、Ad-null+PE组和Ad-Crim1+PE组心室肌细胞大小有差异,见图2。ImageJ软件测量Ad-null组、Ad-null+PE组和Ad-Crim1+PE组心室肌细胞面积分别为(279.76±73.71)μm2、(568.29±84.37)μm2和(396.82±45.64)μm2。PE干预24 h诱导心室肌细胞面积明显增大,该效应可被过表达Crim1抑制(P均<0.001)。
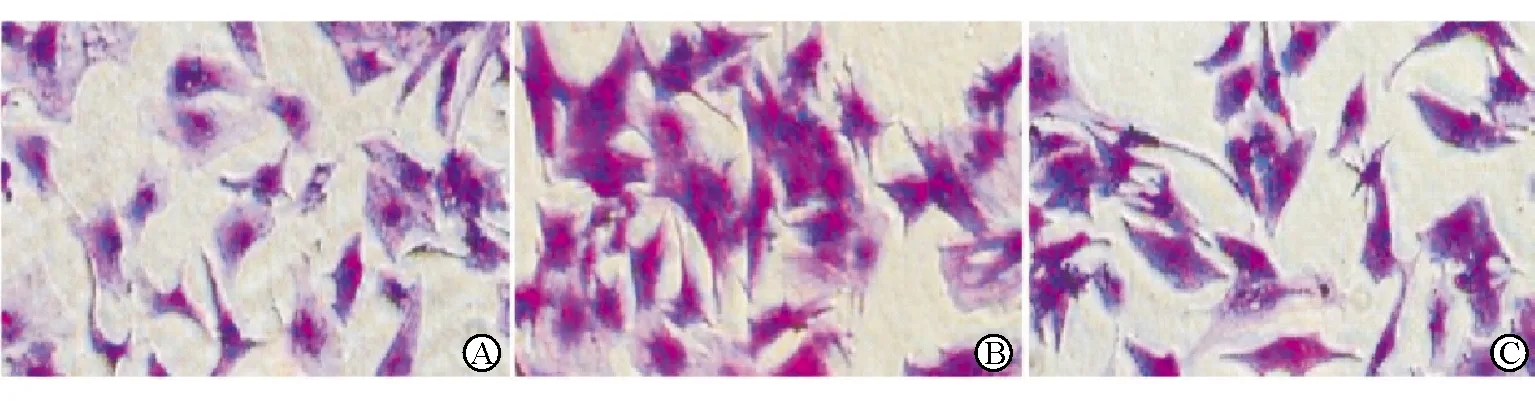
注:A为Ad-null组;B为Ad-null+PE组;C为Ad-Crim1+PE组
2.3 过表达Crim1抑制PE诱导的心室肌细胞Ito改变
在刺激电压为20~70 mV时,Ad-null+PE组细胞Ito电流密度较Ad-null组明显减小,而Ad-Crim1+PE组较Ad-null+PE组明显增大(P<0.05),见图3、表1。
3 讨论
Crim1是Ⅰ型跨膜蛋白,在胎儿生长发育过程中的多个组织器官表达,调控胚胎发育,包括心脏的发育调控[11-15]。Crim1对于胚胎期心脏发育的影响提示其可能参与出生后心肌肥大的病理生理过程,然而,相关证据极少。在培养的牵张刺激肥大乳鼠心室肌细胞和在体腹主动脉结扎肥大大鼠心室肌组织模型中发现,Crim1基因和蛋白表达皆明显下调;给予氯沙坦或替米沙坦干预能显著减轻心室肌细胞肥大和心室肥大,同时上调Crim1基因和蛋白的表达,提示Crim1可能参与对此模型心室肌细胞及心室组织肥大的调控,且Crim1表达对病理性心肌肥大为负性调节作用[6-7]。
本研究发现,给予肾上腺素能α受体激动剂PE干预培养的乳鼠心室肌细胞,导致细胞肥大、细胞Ito电流密度减小;通过重组腺病毒感染心室肌细胞获得Crim1蛋白过表达干预,可明显抑制PE诱导的上述效应,证明Crim1参与心室肌细胞肥大的调控,并证实Crim1参与肥大心室肌细胞离子通道重构的调控。
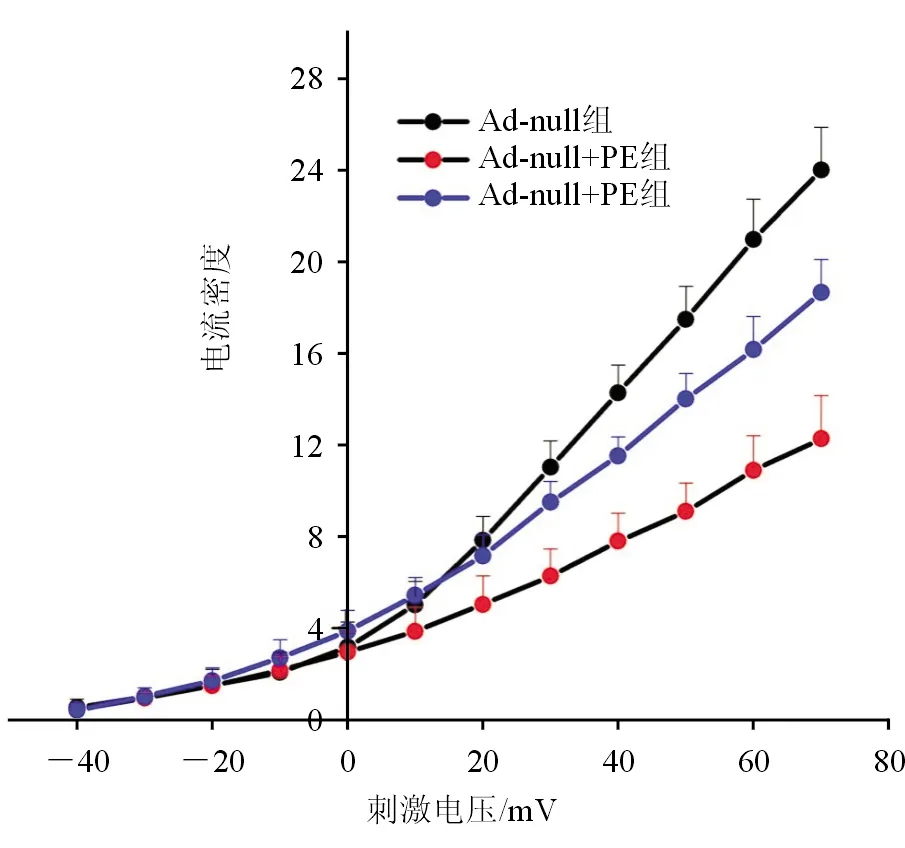
图 3 心室肌细胞瞬时外向钾电流电流的密度

表1 各组不同电压下的瞬时外向钾电流的电流密度比较 (pA/pF,n=10)
Ito是一种快速激活和快速失活的外向钾电流,主要参与动作电位的1期,与心室肌细胞动作电位的形状和时程有关。Ito减小可延缓动作电位1期复极化,减小1期切迹深度,从而影响其他离子通道活性。在器质性心脏病的肥大心室肌中,尤其是合并心力衰竭、心肌损伤时,心室肌细胞编码Ito离子通道的Kv4.3基因及其蛋白表达皆下调,Ito离子通道功能活性减低,导致早期复极异常、复极延迟和APD延长,易发生致命性室性心律失常[16-18]。
本研究结果显示,Crim1过表达对心室肌细胞肥大及伴随的Ito电流密度改变有抑制作用,针对Crim1的干预,有可能对心室肥大及伴随的室性心律失常具有防治作用。
猜你喜欢
中华实用诊断与治疗杂志(2022年1期)2022-08-31
世界科学技术-中医药现代化(2022年2期)2022-05-25
世界科学技术-中医药现代化(2021年12期)2021-04-19
青岛大学学报(医学版)(2020年6期)2020-11-16
解放军医学院学报(2020年12期)2020-03-29
中西医结合心脑血管病杂志(2016年20期)2016-03-01
中国卫生标准管理(2015年17期)2016-01-20
川北医学院学报(2015年5期)2015-12-05
中国当代医药(2015年33期)2015-03-01
西南军医(2014年6期)2014-01-22
