
常染色体隐性遗传念珠状发一家系DSG4基因突变研究
2019-03-23 09:01任英豪陈晨曹瑞祥李昕张江安李小红张贝于建斌孔祥东郑州大学第一附属医院皮肤科45005郑州大学第一附属医院遗传与产前诊断中心45005
中华皮肤科杂志 2019年12期
任英豪 陈晨 曹瑞祥 李昕 张江安 李小红 张贝 于建斌 孔祥东郑州大学第一附属医院皮肤科 45005;郑州大学第一附属医院遗传与产前诊断中心45005
念珠状发(monilethrix)是一种罕见的遗传性皮肤病,表现为特征性毛干发育不良,即正常厚度的椭圆节间有规则的营养不良收缩,呈念珠状外观,薄的节间区域有很高的断裂倾向。临床表现为脱发伴毛囊角化过度和毛囊周围红斑,可仅累及枕部,严重时可累及整个头皮、眉毛及睫毛。本病常染色体显性遗传(OMIM 158000),是由编码毛发角蛋白的基因KRT81、KRT83、KRT86等杂合突变引起,而常染色体隐性遗传是由桥粒芯糖蛋白4(DSG4)基因突变引起[1]。迄今为止,国内外[1-3]仅报道数例DSG4基因突变致常染色体隐性遗传念珠状发。本文报道1例DSG4基因复合杂合突变致常染色体隐性遗传念珠状发。
病历资料
先证者女,3岁,汉族,出生后头皮可见片状毛囊角化性丘疹,无毛发生长。2岁左右开始生长稀疏毛发,粗细不均,间断脱落,触之易断。毛囊角化性丘疹逐渐增多并扩展至双眼睑和枕部。发病以来,精神、饮食、睡眠和大小便正常。患儿系第2胎第2产,足月顺产,无产伤窒息史,母乳喂养,预防免疫接种随当地进行。患儿有1哥哥,表型正常,父母非近亲结婚,毛发密度和分布正常。母亲怀孕期间无服药、接触有害物质及病毒感染史。家族其他成员无类似疾病及其他先天性或遗传病史。本研究经郑州大学第一附属医院科研项目伦理审查委员会批准(2018-YB-05)。
体检:营养良好,身高、体重及智力正常,其他系统检查无明显异常。皮肤科检查:头皮顶部、枕部片状头发稀疏、脱落,残留断发及毛囊角化性丘疹。病发及毛囊角化性丘疹在头皮顶部及枕部明显,部分眉毛、睫毛脱落、折断,长短不一(图1A~1C)。牙齿、指甲、趾甲及汗腺正常。
实验室检查
血常规示白细胞7.6×109/L,红细胞4.39×1012/L,血红蛋白122g/L,血小板411×109/L。血清IgA 0.41 g/L(参考值0.8~5 g/L),血清IgE 126.50 IU/ml(参考值0~60 IU/ml)。尿粪常规、肝肾功能、电解质、空腹血糖、凝血功能、红细胞沉降率、降钙素原、补体C3及C4、25羟基维生素D3、微量元素检测均正常。乙型肝炎病毒、丙型肝炎病毒、梅毒、HIV筛查均未见异常。

图1 念珠状发患儿临床表现及皮肤镜、光镜和电镜检查结果 1A~1C:患儿头皮顶部、颞部片状头发稀疏、脱落,残留断发及毛囊角化性丘疹,部分眉毛脱落、折断,长短不一;1D:皮肤镜下头发毛干粗细不均,长短不一,可见黑点征样改变,头发毛干可见念珠状外观,结节间距较短(×10);1E:光镜下见典型念珠状发(×40);1F:扫描电镜示典型念珠状发(左上角小图,×100),鳞片状毛小皮脱落,毛皮质呈枯树皮样外观(×1 000)

图2 DSG4基因第1、2、10、15、16号外显子实时荧光定量PCR检测结果 先证者和其母亲均第1外显子正常,第2、10、15、16外显子杂合缺失;先证者父亲和健康对照第1、2、10、15、16外显子均正常
皮肤镜及毛发扫描电镜检查
皮肤镜下头发毛干粗细不均,长短不一,可见断发残根,呈黑点征样改变,头发毛干可见念珠状外观,结节间距较短,毛发很短,易于折断(图1D)。光镜下病发呈典型的念珠状外观(图1E)。征得患儿父母知情同意后,取患儿少许毛发,扫描电镜(日本电子株式会社)观察,结果显示:毛发粗细不均,低倍可见典型念珠状发,高倍示病变处鳞片状毛小皮脱落,遗留毛皮质,呈枯树皮样外观(图1F)。
基因检测
征得患儿父母知情同意后采集患儿及其父母血样进行基因测序,以100例健康人外周血作为对照。采用北京迈基诺基因科技有限公司设计的皮肤病相关基因检测包,对先证者皮肤病相关基因外显子区及相邻侧翼序列进行靶向性捕获,使用Illumina Nextseq500测序仪进行高通量测序,平均测序深度为300×,覆盖度为95%。高通量测序结果显示,先证者DSG4基因存在第2~16外显子杂合缺失,以及第6外显子c.574T>C(p.S192p)(NCBI序列号:NM-177986)突变,分别通过实时荧光定量PCR及Sanger测序得到验证。先证者母亲携带DSG4基因2~16号外显子杂合缺失突变,先证者父亲携带DSG4基因c.574T>C(p.S192p)杂合突变。100例健康人均未发现该位点变异。见图2、3。
讨 论
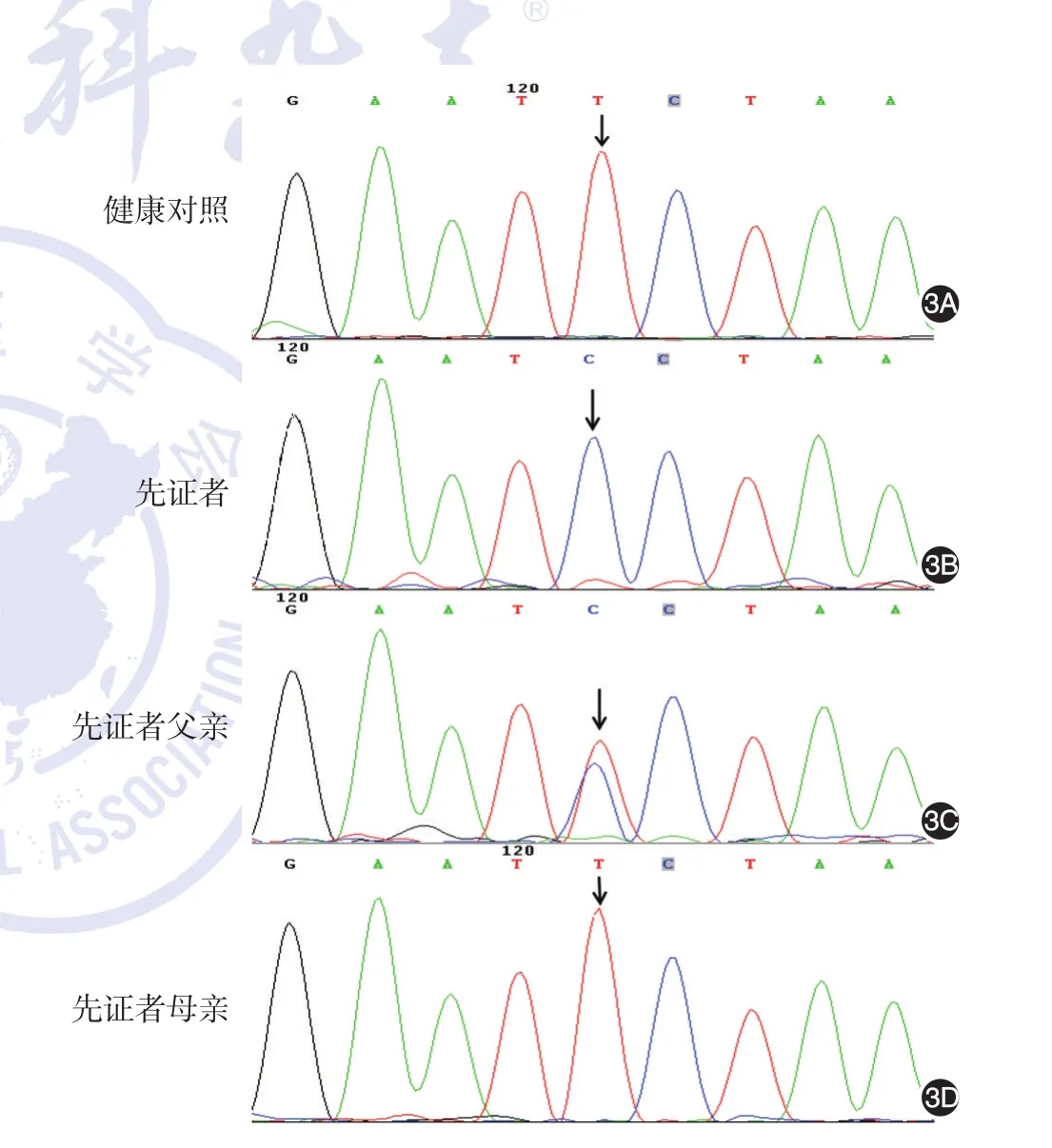
图3 先证者及其父母和健康对照DSG4基因第6外显子574位点正向测序结果 3A:健康对照为T/T纯合峰;3B:先证者表现为c.574T>C(p.S192p)半合子变异:3C:先证者父亲c.574T>C(p.S192p)杂合变异;3D:先证者母亲为正常序列单倍体
桥粒属于钙黏蛋白超家族,由桥粒芯蛋白、桥粒胶蛋白和桥粒斑珠蛋白组成[4]。2003年Whittock和Bower[5]鉴定了一种新的人类DSG,将其命名为DSG4。Kljuic等[6]报道2例局限性常染色体隐性遗传稀毛症(localized autosomal recessive hypotrichosis,LAH),患者临床表现与念珠状发相似,DSG4基因存在5~8号外显子缺失,为致病性突变。2006 年 Shimomura 等[7]、Zlotogorski等[8]和Schaffer等[9]分别报道伴有念珠状发改变的LAH,患者临床表型与念珠状发相似,均发现DSG4基因突变,这表明DSG4基因突变引起的LAH在临床上可以与念珠状发重叠。2009年Bazzi等[10]发现DSG4基因突变导致人毛发减少,体外和体内实验均证明DSG4定位于桥粒,在人类毛囊中具有高度特异的表达模式,对毛干完整性非常重要。
2011年Farooq等[1]报道1例DSG4基因突变致常染色体隐性遗传念珠状发,并发现突变的DSG4对桥粒斑珠蛋白亲和力降低,首次展示了DSG4和桥粒斑珠蛋白的相互作用。2015年Wang等[11]报告1例DSG4基因突变致LAH,患者表现为稀疏毛发及毛囊角化性丘疹,眉毛和睫毛也受损,症状类似念珠状发。王沛等[2]报告1例DSG4基因突变致常染色体隐性遗传念珠状发,患者DSG4基因突变从而形成翻译提前终止密码子,引起DSG4基因翻译提前终止,该突变导致桥粒芯糖蛋白C端结构缺失,影响DSG和桥粒斑珠蛋白的连接,导致细胞间连接系统缺陷,从而出现念珠状发,并指出LAH患者的DSG4基因突变报道都为纯合突变,而念珠状发患者几乎均为杂合突变,与我们本次报道DSG4基因杂合突变导致念珠状发一致。王沛等[2]和Wang等[11]均认为DSG4基因突变导致的LAH和念珠状发可能为同一种疾病,但还需更多病例报告和突变研究来证实。本文先证者携带DSG4基因c.574T>C(p.S192p)(NM-177986)突变,与既往文献[7]报道相同,该位点在人类DSG中完全保守,该突变为致病突变。同时,本文先证者DSG4基因2~16号外显子缺失,检索国内外文献未发现有相同缺失突变的案例报道。既往文献[6,12]报道LAH患者DSG4基因5 ~8号外显子缺失,产生类似念珠状发表现。考虑到患者DSG4基因存在大片段缺失突变,推测为致病性变异。综上推测本例患儿携带的DSG4基因点突变和缺失突变产生异常的DSG4蛋白,导致细胞间连接系统缺陷,影响相邻细胞间黏附,从而引起毛发角蛋白表达异常,患儿出现念珠状发表型。
2016年Muhammad等[12]对6个LAH直系血亲家族的DSG4基因序列分析发现,6个家族的受影响个体中都发生了5~8号外显子缺失突变。这些家族的其他13种突变中,2种突变引起念珠状发,3种导致念珠状发和LAH并存,剩下的8种导致LAH,推测DSG4基因突变的表型变异可能与不同修饰基因的作用有关。
常染色体隐性遗传念珠状发和LAH的致病基因均为DSG4,临床表现相似,表现为脱发、毛发稀疏等,差异为念珠状发患者更容易出现角化过度的毛囊丘疹和毛囊周围红斑,毛干呈典型念珠状发样外观。依据典型的临床特征、家族史、实验室检查、光镜、皮肤镜、扫描电镜检查和基因检测,本例患儿常染色体隐性遗传念珠状发诊断明确。
猜你喜欢
临床输血与检验(2022年3期)2022-06-22
种子(2021年3期)2021-04-12
阅读时代(2020年11期)2020-09-10
诊断学(理论与实践)(2020年1期)2020-04-28
郑州大学学报(医学版)(2019年3期)2019-06-03
校园英语·下旬(2017年7期)2017-07-14
现代家庭(2017年4期)2017-04-15
科技视界(2016年27期)2017-03-14
中学生理科应试(2016年7期)2016-05-14
小小说月刊·下半月(2010年8期)2010-05-14
