
基质固相分散-快速溶剂萃取-GC/MS法同时测定土壤中有机氯农药和多环芳烃
2019-03-13 06:14王伟
中国环境监测 2019年1期
王 伟
1.太原市环境监测中心站,山西 太原 030002 2.太原理工大学环境科学与工程学院,山西 太原 030024
有机氯农药(OCPs)和多环芳烃(PAHs)是具有致癌、致畸、致突变效应的持久性有机污染物[1]。六六六、滴滴涕(DDT)、滴滴滴(DDD)等是典型的有机氯农药,曾是广泛使用的杀虫剂,主要用于农业、环境以及卫生方面,但由于该类农药具有半衰期长、不易分解和亲脂性等特点,使用中易造成富集,从而污染环境[2-4]。多环芳烃主要来源是各种矿物燃料(如煤)、石油以及垃圾、木材等的不完全燃烧,由于其低溶解性及脂溶性,比较容易进入生物体内,进而通过“食物链”或其他途径进入到人体内,对人体健康构成危害[5-6]。
测定土壤中OCPs和PAHs的传统方法是索氏提取[7],该方法存在提取时间长、溶剂用量大[8]、提取液处理过程复杂等缺点。快速溶剂萃取(ASE)[9-10]是近年来发展起来的在高温高压下快速提取固体或半固体样品中目标物的一种新方法,该方法操作简单,提取速度快,目标物的提取效率高。但同时对于一些杂质的提取效率也同样得到增强,使得提取液中含有大量杂质,提取液的净化步骤较多,过程繁琐费时。采用基质固相分散(MSPD)[11-12]辅助ASE提取,土壤样品在萃取池中提取与净化一步完成,改变了传统的样品先提取后净化的流程,从而大大提高了土壤样品的分析效率[13-14]。本实验采用硅藻土结合弗罗里硅土作为分散剂辅助ASE提取土壤中8种有机氯农药和16种多环芳烃,提取液只需脱水浓缩后即可通过GC/MS法进行测定,方法快速、简单。
1 实验部分
1.1 仪器与试剂
气相色谱-质谱联用仪,7890B-5977A型,美国;色谱柱,DB-5ms石英毛细管柱,30 m×250 μm×0.25 μm;加速溶剂萃取仪,ASE350型,配34 mL不锈钢萃取池,美国;全自动定量浓缩仪,DryVap型,美国;天平,万分之一天平,AB204-E型。

1.2 土壤样品制备
采集的土壤样品混均后,将其中混杂的石子、树枝等杂物除去,研碎后过尼龙筛,过筛后的样品混匀备用。如暂不分析可保存在-18 ℃冷冻箱中。
1.3 土壤样品处理
1.3.1 土壤样品提取
准确称取10.0 g土壤样品置于研钵中,加入1.0 g硅藻土,研磨混匀,再加入5.0 g弗罗里硅土,充分研磨混匀,转入底部铺有玻璃纤维滤膜和5.0 g弗罗里硅土的34 mL不锈钢萃取池中,盖好顶盖并拧紧,将萃取池垂直放入快速溶剂萃取仪样品盘后运行仪器进行提取,提取溶剂为丙酮-正己烷(体积比1∶1)。快速溶剂萃取仪的提取条件:加热温度100 ℃;加热时间5 min;静态萃取时间5 min;循环次数2次;冲洗体积60%;吹扫时间100 s。
1.3.2 样品提取液浓缩
收集瓶中加入适量无水硫酸钠脱水后,将提取液全部转入DryVap浓缩杯中,再用10 mL正己烷分3次冲洗收集瓶,洗液同样转入浓缩杯中,在全自动浓缩仪中浓缩至0.9 mL左右,再转入氮吹管中氮吹浓缩至约0.5 mL,加入内标溶液后,用正己烷定容至1.0 mL,通过GC/MS进行测定。
1.4 气相色谱条件
进样方式为不分流进样;进样体积为1.0 μL;进样口温度280 ℃;程序升温时起始温度50 ℃,保持2.0 min,以20 ℃/min升温至180 ℃,并保持5.0 min,再以10 ℃/min升温至250 ℃,以5 ℃/min升温至300 ℃保持5.0 min;载气流速1.0 ml/min,恒定流速。
1.5 质谱条件
电离方式EI,70 eV;离子源温度230 ℃;四级杆温度150 ℃;传输线温度300 ℃;溶剂延迟5 min;扫描方式为全扫描Scan定性,质量扫描范围45~450 amu;选择离子SIM定量。
2 结果与讨论
2.1 目标化合物的分离结果
采用全扫描Scan方式,对8种有机氯农药和16种多环芳烃以及2种内标物进行定性。标准溶液中目标化合物和内标物的全扫描总离子流(TIC)色谱图见图1。
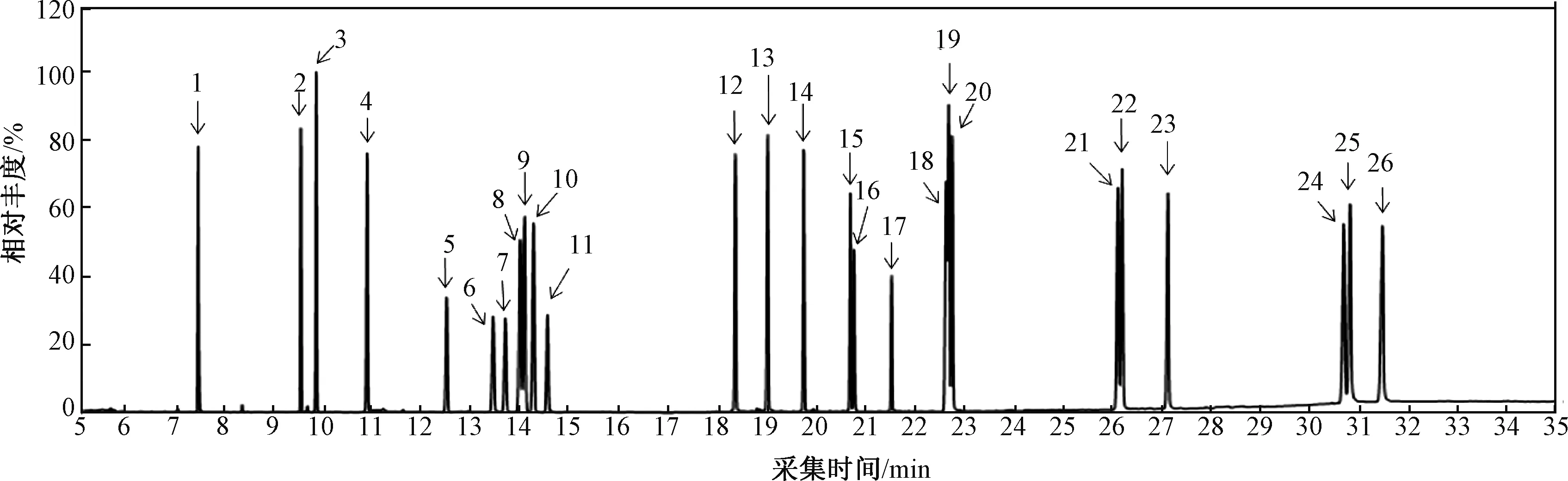
1.萘;2. 苊烯;3. 苊;4. 芴;5. α-六六六;6. β-六六六;7. γ-六六六;8. 氘代菲(内标1);9. 菲;10. 蒽;11. δ-六六六;12. 荧蒽;13. 芘;14. p,p′-DDE;15. p,p′-DDD;16. o,p′-DDT;17. p,p′-DDT;18. 苯并(a)蒽;19. 氘代(内标2);20. ;21. 苯并(b)荧蒽;22. 苯并(k)荧蒽;23. 苯并(a)芘;24. 茚并(123-c,d)芘;25. 二苯并(a,h)蒽;26. 苯并[g,h,i]苝图1 目标化合物和内标物全扫描TIC色谱图Fig.1 The full scan TIC of target compounds and internal markers
在这些目标化合物中,有多种同分异构体,物质对的定性离子和定量离子都相同,只有分离好才能实现结果测量准确。由图1可见,在该色谱条件下,目标化合物以及内标物得到较好的分离,能够满足实验要求。
为降低目标物的检出限,采用选择离子方式SIM进行定量。图2为目标化合物和内标物的选择离子扫描TIC色谱图。各目标化合物和内标物的保留时间、定量离子和定性离子见表1。

1.萘;2. 苊烯;3. 苊;4. 芴;5. α-六六六;6. β-六六六;7. γ-六六六;8. 氘代菲(内标1);9. 菲;10. 蒽;11. δ-六六六;12. 荧蒽;13. 芘;14. p,p′-DDE;15. p,p′-DDD;16. o,p′-DDT;17. p,p′-DDT;18. 苯并(a)蒽;19. 氘代(内标2);20. ;21. 苯并(b)荧蒽;22. 苯并(k)荧蒽;23. 苯并(a)芘;24. 茚并(123-c,d)芘;25. 二苯并(a,h)蒽;26. 苯并[g,h,i]苝图2 目标化合物和内标物选择离子扫描TIC色谱图Fig.2 The SIM TIC of target compounds and internal markers

序号化合物定量离子定性离子保留时间/min线性方程相关系数1萘128127、1297.398Y=1.702 2X-0.068 50.998 32苊烯152151、1539.452Y=0.747 0X-0.009 20.999 63苊154153、1529.763Y=0.842 8X-0.003 40.999 64芴166165、16710.777Y=0.837 0X-0.002 50.999 75α-六六六183181、10912.367Y=0.146 7X-0.003 50.999 46β-六六六181183、10913.258Y=0.115 4X-0.002 60.999 27γ-六六六183181、10913.522Y=0.139 9X-0.006 50.999 28氘代菲(内标1)188189、16013.823//9菲178179、17613.909Y=1.366 0X-0.011 50.999 610蒽178179、17614.097Y=0.629 3X-0.019 80.999 411δ-六六六183181、10914.392Y=0.111 9X-0.003 40.999 212荧蒽202200、20318.199Y=0.791 1X-0.013 30.999 213芘202200、20318.848Y=1.028 4X-0.055 10.998 314p,p′-DDE246248、17619.594Y=0.330 8X+0.026 40.999 515p,p′-DDD235237、16520.529Y=0.261 5X+0.028 80.999 416o,p′-DDT235237、16520.604Y=0.206 3X+0.002 70.999 817p,p′-DDT235237、16521.361Y=0.148 9X+0.015 40.999 018苯并(a)蒽228226、22922.438Y=0.514 0X+0.025 60.998 319氘代艹屈(内标2)240236、23822.487//20艹屈228226、22922.557Y=1.189 5X+0.000 80.999 821苯并(b)荧蒽252253、25025.891Y=0.564 7X+0.033 90.998 222苯并(k)荧蒽252253、25025.966Y=0.752 7X+0.002 30.997 823苯并(a)芘252253、25126.888Y=0.377 5X+0.010 10.999 324茚并(123-c,d)芘276277、27530.411Y=0.349 6X+0.004 80.997 525二苯并(a,h)蒽278276、27930.547Y=0.483 7X-0.001 60.998 426苯并[g,h,i]苝276275、27431.174Y=0.689 0X-0.016 10.997 7
注:“/”表示内标物不进行线性回归。下同。
2.2 分散剂的选择
在基质固相分散萃取中,分散剂起着分散、支持、吸附和净化的作用。本实验分别考察了硅藻土、弗罗里硅土以及硅藻土结合弗罗里硅土作为分散剂对土壤的分散和净化效果。结果表明,硅藻土对土壤有良好的分散性,但对土壤提取液的净化能力有限;弗罗里硅土对土壤提取液有较强的净化能力,但对较潮湿的新鲜土壤的分散能力较差;硅藻土结合弗罗里硅土则能够较好实现土壤分散和净化效果。因此,本实验使用硅藻土结合弗罗里硅土作为土壤的分散剂。
2.3 硅藻土结合弗罗里硅土辅助ASE萃取
在萃取池底部铺一层弗罗里硅土,再将土壤混合样品置于弗罗里硅土上层,如图3所示。这样,提取时顺着溶剂的流向,提取液中的脂肪、色素等的干扰物会被弗罗里硅土吸附留在萃取池内,而目标化合物则随提取液进入收集瓶中,从而达到一步提取和净化的效果。提取液只需脱水浓缩后即可通过GC/MS进行检测,与传统的先萃取再净化的方法相比,能明显节省时间、人力,减少试剂及材料的消耗。

图3 硅藻土结合弗罗里硅土辅助ASE萃取图Fig.3 Diatomaceous earth and Florisil-assisted ASE extraction system
考察了不同用量的弗罗里硅土对净化效果的影响。在污染较轻土壤和污染较重土壤中分别加入3、4、5 g弗罗里硅土,对提取液进行对比。结果发现,分别加入3、4、5 g弗罗里硅土的污染较轻土壤的提取液均为无色透明,杂峰较少;而污染较重的土壤随弗罗里硅土加入量的增加,提取液的颜色逐渐变浅。结果表明,对未受污染或污染较轻的农田土壤等,可适当减少弗罗里硅土的用量;而对污染较重的土壤需适当增加弗罗里硅土用量,以达到更好的净化效果。
2.4 绘制校准曲线

2.5 方法检出限
依据检出限的计算方式,连续分析7个接近于检出限浓度的空白加标样品,计算其标准偏差(S),按MDL=S×t(n-1,0.99)(t6,0.99=3.143)计算方法检出限。本实验以石英砂为空白基体,加入六六六、DDT混合溶液和多环芳烃标准溶液,配制7个有机氯农药质量分数为5 μg/kg、多环芳烃质量分数为2 μg/kg的空白加标样品,经提取和浓缩后上机测定,计算得出各化合物组分的方法检出限,结果见表2。
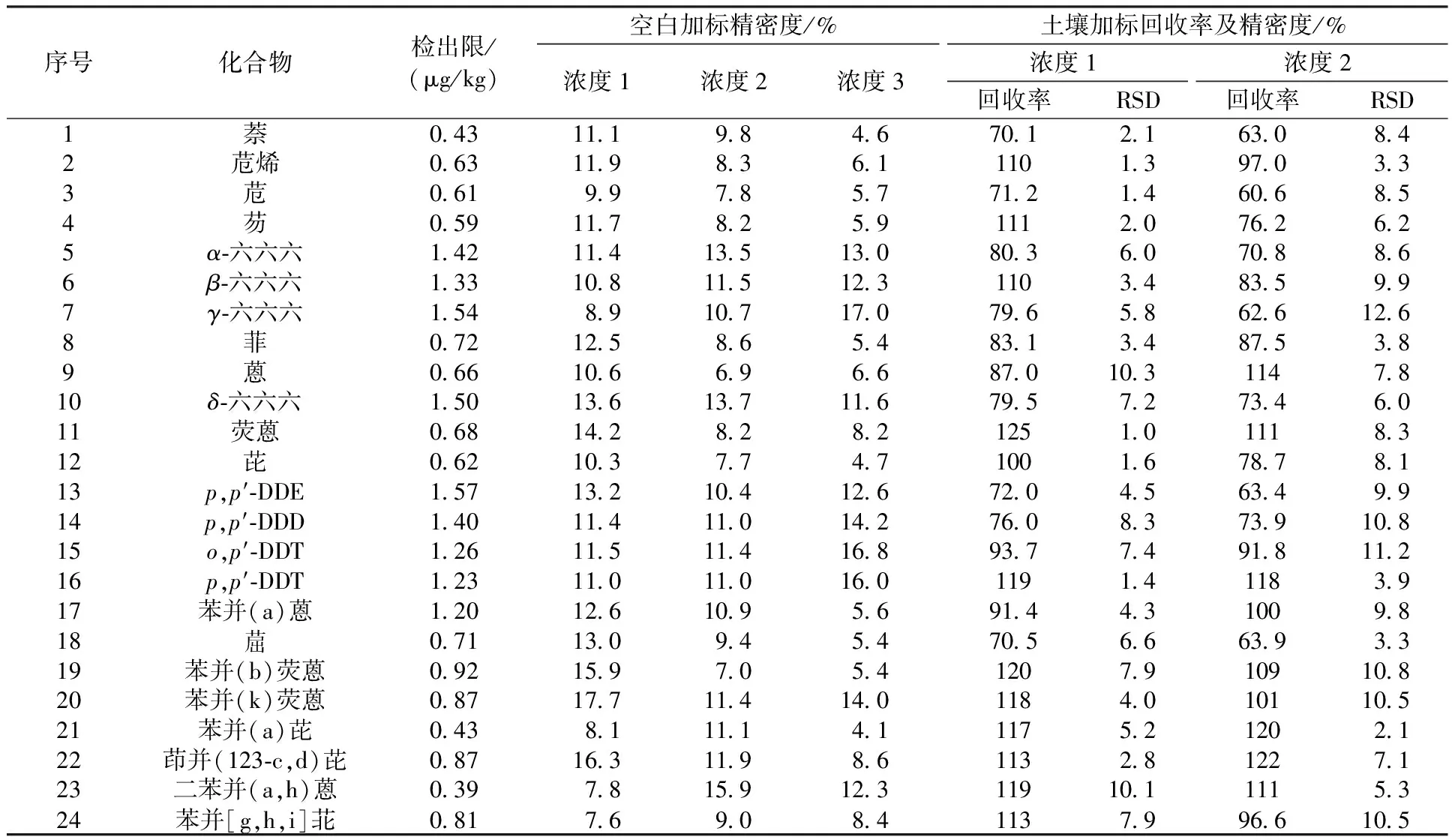
表2 各化合物组分的方法检出限、精密度及加标回收率Table 2 The detection limit, precision and recovery of compounds
2.6 方法精密度及准确度
本实验以石英砂为空白基体,加入六六六、DDT混合溶液和多环芳烃标准溶液,制成3组不同加标浓度的空白加标样品。浓度1:有机氯农药5 μg/kg,多环芳烃2 μg/kg;浓度2:有机氯农药20 μg/kg,多环芳烃10 μg/kg;浓度3:有机氯农药40 μg/kg,多环芳烃16 μg/kg,经提取和浓缩后上机测定,计算得出各组分的精密度,结果见表2。由表2可知,空白加标样品的相对标准偏差(RSD)小于20%,精密度良好。
本实验以实际土壤样品作为基体,加入六六六、DDT混合溶液和多环芳烃标准溶液,制成2组不同加标浓度的土壤加标样品。浓度1:有机氯农药20 μg/kg,多环芳烃10 μg/kg;浓度2:有机氯农药40 μg/kg,多环芳烃16 μg/kg,经提取和浓缩后上机测定,计算得出各组分的加标回收率,结果见表2。由表2可知,实际土壤样品的加标回收率为60.6%~125%,相对标准偏差小于15%。
2.7 对土壤质控样品的测定
本实验对SQCO-003土壤有机氯农药质控样品和017土壤多环芳烃质控样品进行了测定,结果见表3。由表3可知,各组分测定结果均在质控范围内,本方法准确可靠。
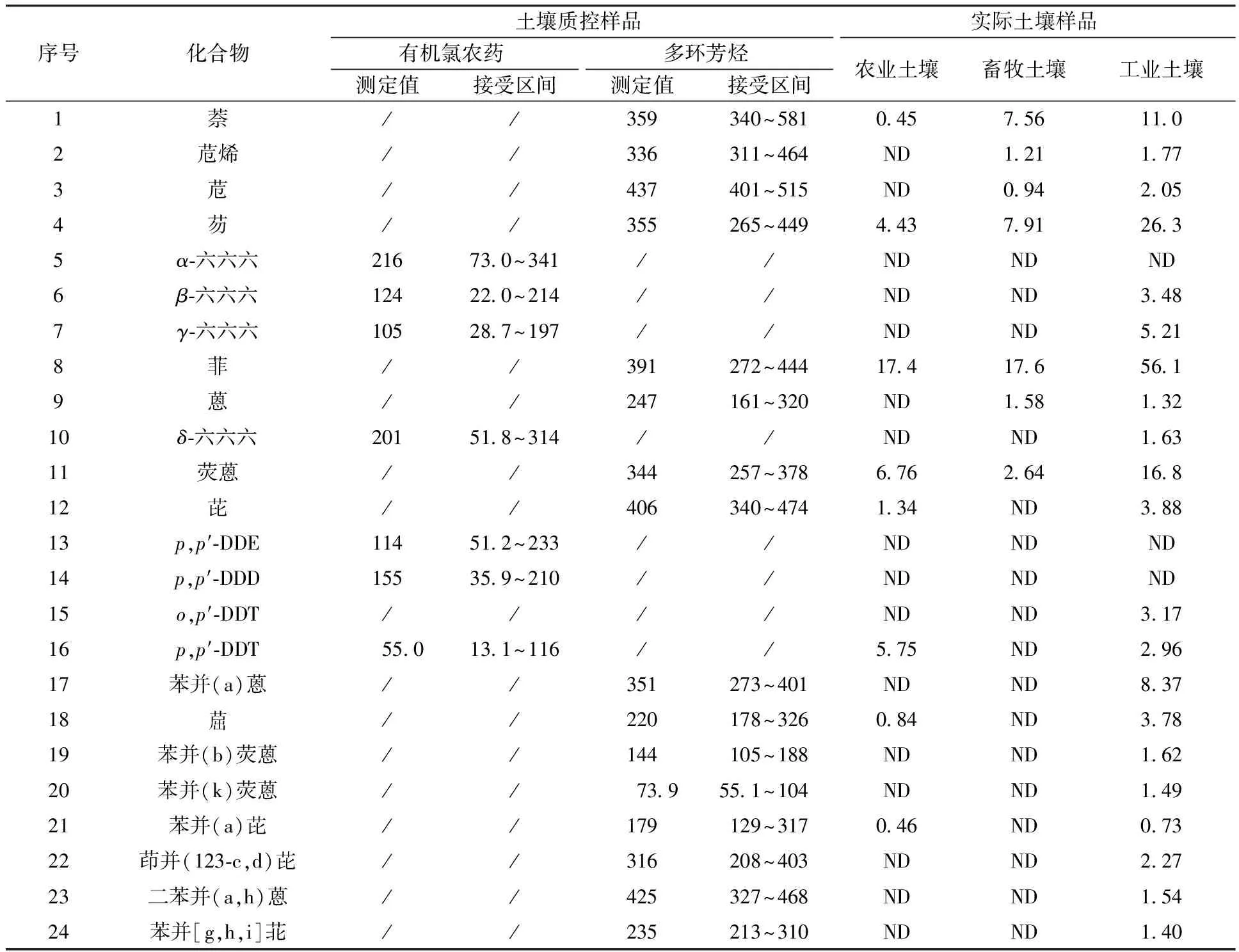
表3 土壤质控样品和实际土壤样品测定结果Table 3 The results of soil QC sample and actual sample μg/kg
注:ND表示未检出。
2.8 对实际土壤样品进行测定
应用本方法对城市周边典型的农业、畜牧业、工业用地的实际土壤样品进行测定,结果见表3。由表3可知,有机氯农药和多环芳烃在农业、畜牧业、工业用地土壤中均有不同程度的检出,其中该工业用地土壤污染物检出较为显著。
3 结论
通过实验建立了基质固相分散-ASE提取-GC/MS法同时测定土壤中8种有机氯农药和16种多环芳烃的方法,本方法在线性范围内具有良好的相关系数、较低的检出限以及较好的精密度、准确度,通过对土壤有机氯农药质控样品和土壤多环芳烃标准样品以及实际土壤加标样品的测定,说明本方法准确可靠。本方法操作简单、自动化程度高,土壤样品在萃取池中提取与净化一步完成,缩短了样品的处理步骤和时间,从而提高了样品分析效率,能够实现土壤样品的快速检测,适合大批量样品的处理分析。
猜你喜欢
水泵技术(2022年3期)2022-08-26
石油沥青(2021年4期)2021-10-14
科学家(2021年24期)2021-04-25
石油化工技术与经济(2021年6期)2021-01-12
食品安全导刊(2020年30期)2020-12-03
食品安全导刊(2020年23期)2020-12-03
食品安全导刊·中旬刊(2020年8期)2020-09-22
扣篮(2019年3期)2019-03-25
食品安全导刊(2018年1期)2018-02-11
文学少年(小学版)(2016年11期)2016-12-17
