
洋葱伯克霍尔德菌复合群部分菌株基于基因组的物种水平重鉴定
2019-02-28 03:26:38周江林孔娜张琪胡明达周静岳俊杰任洪广靳远梁龙
生物技术通讯 2019年6期
周江林,孔娜,张琪,胡明达,周静,岳俊杰,任洪广,靳远,梁龙
军事科学院 军事医学研究院 生物工程研究所,北京100071
洋葱伯克霍尔德复合群(Burkholderia cepa⁃ciancomplex,Bcc)是以康奈尔大学植物病理学家Burkholder 命名的一个细菌复合群,它的模式菌就是洋葱伯克霍尔德菌(B.cepacia),由Burkholder 于1949年首次发现,因其引起洋葱表皮腐烂而命名为“洋葱(cepacia)”[1],最初被称作洋葱假单胞菌(Pseudomonas cepacia),随后在1992 被重新分类为β变形杆菌纲、伯克霍尔德目、伯克霍尔德科、伯克霍尔德属而称为洋葱伯克霍尔德菌[2]。Bcc是一群非葡萄糖发酵剂的好氧革兰阴性杆菌,包括至少22种不同的菌种[3]。这些菌种系统发生关系密切相关,通常使用洋葱伯克霍尔德菌的recA基因序列进行种鉴定[4]。这些种当中有9个先前被命名为以下伯克霍尔德基因型:B.cepacia(ge⁃nomovar Ⅰ)、B.multivorans(genomovar Ⅱ)、B.ceno⁃cepacia(genomovar Ⅲ)、B.stabilis(genomovar Ⅳ)、B.vietnamiensis(genomovarⅤ)、B.dolosa(genomovarⅥ)、B.ambifaria(genomovar Ⅶ)、B.anthina(genom⁃ovar Ⅷ)、B.pyrrocinia(genomovar Ⅸ),随后不断被确认并命名为新的物种。Bcc 广泛存在于自然水源、土壤和其他潮湿的环境中,它们可以在这些环境存活达数月之久[5-6],现有证据显示环境可能是新型Bcc 感染的源头[7]。自20世纪50年代首次报告人感染洋葱伯克霍尔德菌患病以来[1],现在已是人群中流行程度仅次于ESKAPE病原体(肠球菌、金黄色葡萄球菌、克雷伯菌、不动杆菌、铜绿假单胞菌、肠杆菌)的致病菌。Bcc 频繁地从囊性纤维化患者的痰液当中分离出来,由于其抗生素耐药性和治疗困难,往往会加速囊性纤维化患者肺功能衰竭并增大死亡率[8-9]。同时,Bcc 也会导致慢性肉芽肿病患者的吞噬细胞无法产生活性氧,从而造成致命性疾病[10-11]。近年来不断增加的Bcc 导致的院内感染报告使得这些细菌被认为是紧急医院病原体[12]。
由于Bcc细菌群体高度的表型和基因型相似性,准确地识别鉴定这些菌种比较困难,而错误的病原菌鉴定可能会导致选择不恰当的治疗方法与用药。常规生化反应等表型检测方法无法有效鉴定Bcc菌种,特别是对于很多Bcc菌株甚至会报告不同的科属[3]。16S rRNA 被广泛用于细菌鉴定,但Bcc 中不同种的16S rRNA基因序列相似性高达98%~100%,无法有效鉴别Bcc菌种[13]。Bcc的recA基因被认为具有较高的分辨能力,能够有效区分Bcc 群内菌种[4],并且特异性的recA基因引物对通常被临床上用于recA基因的扩增与Bcc的鉴定[14]。为了克服单基因分辨力较低等缺陷,基于7个看家基因的多位点序列分析(multilo⁃cus sequence analysis,MLSA)被开发出来并作为最有效的分类工具用于Bcc菌群[15]。此外,基于细菌全基因组信息的基因组平均核酸一致性(av⁃erage nucleotide identity,ANI)比较为物种的全面划分提供了新的解决途径,并且ANI值已成为现代细菌分类学中物种分界的金标准,95%~96%的ANI值被提出且普遍接受为细菌物种分界线[16-17]。
随着全基因组测序技术的发展和测序成本的降低,越来越多的Bcc基因组被测序并完整拼接,为生物信息学分析提供了良好的数据基础。本研究使用了所有目前可用的Bcc细菌全基因组序列数据,基于多种方法进行了系统发生和分类现状分析,初步纠正了一些Bcc菌株当前错误的种鉴定。我们的研究结果提示许多GenBank 公共数据库中的Bcc菌株可能存在错误的种鉴定,需要进行深入分析和纠正。
1 材料与方法
1.1 菌株基因组序列数据筛选与下载
从GenBank基因组数据库(https://www.ncbi.nlm.nih.gov)选取72株注释为洋葱伯克霍尔德复合群内菌种的全基因组序列数据(截至2019-04-25)进行分析,其中模式株或代表株基因组9个。这些基因组按照Parks 等所述方法进行去重[18],惟一例外是本研究中去重是基于基因组序列ANI 而不是原文中bac120 比对的两两之间的平均氨基酸一致性(average amino acid identity,AAI)。用checkM 工具评估下载的基因组数据的完整度(completeness)和污染程度(contamination)[19],定义基因组质量(quality)为完整度-5×污染程度[18],去除所有基因组数据质量值低于50的菌株,去重之后共得到62株细菌基因组进行后续分析。选取复合群外的菌株B.pseudomalleiK96243、B.glumaeLMG2196和B.oklahomensisC6786 作 为 外群。
1.2 系统发生分析
对于recA单基因系统发生分析,选取新洋葱伯克霍尔德菌的模式株J2315(=LMG1665)的recA基因全长序列(NCBI 核酸序列访问号为ALK16523.1)作为搜索请求,用blastn 获取所有样本Bcc菌株基因组中的全长recA基因,所有序列使用muscle 工具进行多序列对齐[20],比对结果用trimAl 工具进行剪裁[21],移除所有gaps 超过50%的位点,比对结果共有1071个核苷酸位点。使用MEGA-X 进行1000 次bootstrap 构建最大似然系统发生树[22],模型方法为General Time Reversible model,位点替换速率模型为Gamma Distributed With Invariant Sites(G+I)。
对于多基因位点系统发生分析,使用7个管家基因(atpD、gltB、gyrB、recA、lepA、phaC和trpB)进行Bcc菌株系统发生分析。选取新洋葱伯克霍尔德菌的模式株J2315(=LMG1665)的序列作为搜索请求进行基因位点序列获取,从PubMLST 数据库(https://pubmlst.org/bcc/)下载J2315株的7个位点序列[23],分别为atpD(443 bp)、gltB(400 bp)、gyrB(454 bp)、recA(393 bp)、lepA(395 bp)、phaC(385 bp)、trpB(301 bp)。分别以下载的7个基因位点序列作为请求,用blastn 从每一个样本基因组中获取其对应的基因序列,并用muscle 工具做多序列对齐[20],比对结果alignment 用trimAl 工具进行剪裁[21],移除所有gaps 超过50%的位点。之后将7个基因位点的多序列比对结果用AMAS软件进行拼接[24],拼接好的超矩阵按上述方法使用MEGA-X 构建最大似然系统发生树[22]。构建好的系统发生树用在线工具iTOL(https://itol.embl.de)进行标注和展示[25]。
1.3 基因组比较
用fastANI 工具计算所有样本菌株任意2个基因组之间的ANI值,以及Bcc菌株基因组与外群菌株基因组直接的ANI值[26]。对于同一个种的某一群基因组,若其中任意2个基因组之间的ANI值大于或等于99.95%,则被认为是高度相似而冗余的菌株,从中挑选一个基因组作为该群的代表株,优先选择参考株(reference strain)或代表株(representative strain),若不存在时随机挑选一个完整基因组作为该群的代表株。
2 结果
2.1 recA基因序列分析
62株Bcc的recA基因平均长度为1072.25 bp,最长的recA基因为2株B.ambifaria菌的1080 bp,最短的recA基因为4株B.cenocepacia菌的1070 bp;序列相似度为94.118%~100%,平均95.898%(数据未示)。基于recA序列的系统发生树如图1,B.ubonensis(6株)、B.vietnaminensis(6株)、B.con⁃taminans(2株)的所有菌株都在同一个进化分支上,B.multivorans除了1株外其他6株也均在同一进化分支上且分支长度较短,说明这几株的分类和鉴定相对比较清晰和可靠。新洋葱伯克霍尔德(B.cenocepacia)菌株主要分布在2 大枝上,分别对应图1 中所标示的基因型ⅢA和ⅢB,有个别新洋葱伯克霍尔德菌零星分布在其他进化分支上,需要对照多位点序列系统发生树进一步分析。值得注意的是,我们发现有5株细菌的分布位置与其当前鉴定种的进化分支差异较大,在图1 中黑体加粗表示,其种属情况需要进一步分析。
2.2 MLSA 进化分析
为了进一步分析Bcc菌株的系统发育和分类情况,我们做了MLSA 分析。62株Bcc菌株都成功找到了所有7个看家基因的同源保守位点,多序列对齐后串联长度为2771 bp[atpD(443 bp)、gltB(400 bp)、gyrB(454 bp)、recA(393 bp)、lepA(395 bp)、phaC(385 bp)、trpB(301 bp)],基于串联等位基因序列的最大似然系统发生树如图2。首先,recA基因进化树上处于单一分支的几个种在图2的串联看家基因片段进化树上依然处于单独的进化分支,因此这几个Bcc菌种可能是单系发生的(monophytic),并且其菌株的鉴定和分类情况相对较好。其次,图2 再次确认了新洋葱伯克霍尔德菌至少分2个基因型ⅢA、ⅢB,这与以往的研究结果一致。第三,对于recA基因进化树上5个分布位置异常的菌株(图1),我们发现在基于MLSA 进化树上分布位置也显示与其注释菌种所在进化分支存在较大差异(图2),且这5株菌在2个进化树上的分布位置相互吻合,再次提示我们这几株菌的种属鉴定可能存在问题。
2.3 基因组平均核酸一致性计算与比较结果
为了进一步确认这5株菌的种属信息,我们计算比较了其各自与注释物种、本文进化树上分支所在菌种的参考株或代表株的ANI值,结果见表1。可以看到,菌株DDS 7H-2的当前鉴定菌种是B.cepacia,但是其在系统发生树上处于B.ceno⁃cepacia基因型ⅢA 所在的进化分支,与B.cenocepa⁃cia模式株J2315基因组的ANI值为98.949%,远远大于其与当前鉴定菌种B.cepacia模式株ATCC 25416的ANI值91.9433%,也显著大于细菌物种分界的ANI 阈值95%[17]。结合ANI值比较和进化树分析,我们有足够理由认为菌株DDS 7H-2 实际上是一株新洋葱伯克霍尔德菌(B.cenocepacia),且其基因型为ⅢA。同样的分析,我们可以纠正菌株FDAARGOS 496、LO6、DWS 37E-2、DDS 22E-1的菌种鉴定,详细信息见表2。

图1 62株Bcc菌株基于recA基因序列的系统发生树
3 讨论
准确鉴定细菌种属信息非常重要,尤其是当这些细菌会引起人或动物感染发病并且需要选择对应治疗方案的时候,准确可靠的鉴定信息对于应对传染病暴发和疾病防控至关重要。洋葱伯克霍尔德菌复合群由于其广泛的分布环境和经常性感染人群,特别是在肺纤维化患者间的相互传播给人们带来较大的生命财产损失,引起了研究者的关注。该复合群已知菌种至少有22种,各个种的外形和生化特性比较相似,16S rRNA 序列相似度也非常高,临床上常规生化方法和广泛使用的16S rRNA基因分类方法不能准确有效地分辨各个菌种[3],甚至会产生错误的鉴定注释信息,从而对后续治疗方案选择带来误导;另一方面,现有菌株的测序结果往往会上传到公共序列数据库,这在方便研究人员基于大规模序列数据集综合分析的同时,也可能会由于之前不可靠方法鉴定的菌株注释信息给研究者的分析结果带来偏差甚至错误。
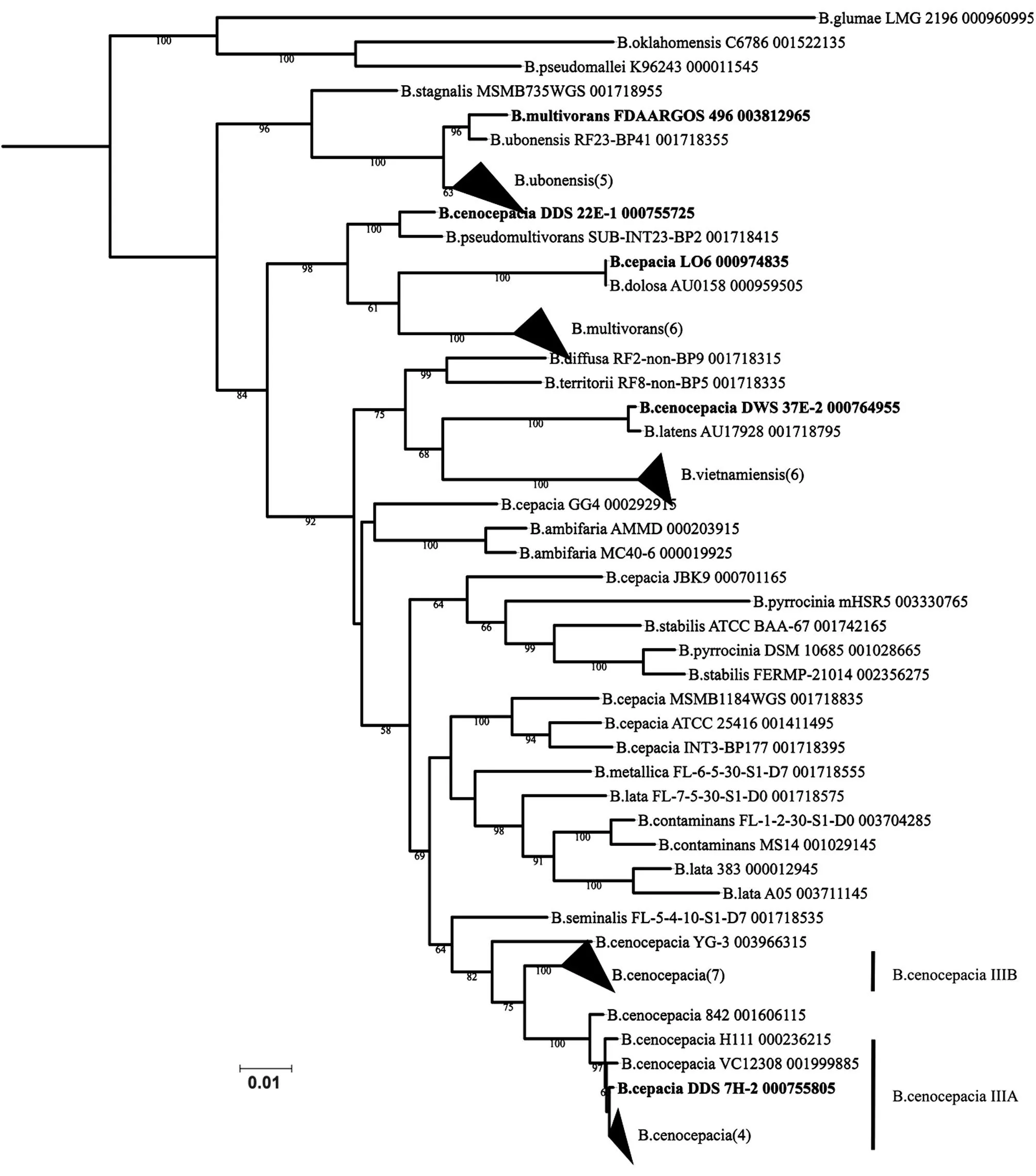
图2 62株Bcc菌株基于7个看家基因片段串联序列的系统发生树

表1 当前种鉴定存在疑问的菌株与Bcc相关菌种基因组ANI值比较(%)

表2 当前种鉴定存在疑问的菌株信息与种鉴定纠正
本研究中我们首先通过构建recA单基因系统发生树,发现有5株细菌在进化树上分布位置与其本身注释菌种所在分支不同并且进化距离较大,而与其他种的细菌分布在一个进化分支且分支长度较小,这提示我们这几株细菌在公共数据库里的分类注释可能存在异常。进一步,我们用7个看家基因片段串联进化分析和细菌全基因组ANI值比较等多种方法,综合分析了当前Bcc细菌的分类情况,结果显示这5株菌当前的菌种注释存在错误,并且根据多种方法更新纠正了这5株Bcc菌株的种鉴定信息。我们的分析和结果表明细菌全基因组信息的ANI值比较与MLSA 合作分析可以良好地鉴定Bcc菌种并避免可能的种鉴定错误,临床上应该以这2种方法的鉴定结果作为标准,避免仅仅依靠单个基因而对Bcc 复合群的细菌做出种鉴定;同时提示当前公共数据库中可能存在许多种鉴定错误的Bcc菌种,这些序列注释信息需要研究人员进一步分析和纠正。
猜你喜欢
南华大学学报(自然科学版)(2021年5期)2021-11-17 04:40:36
实验与检验医学(2021年2期)2021-05-19 03:52:24
Acta Mathematica Scientia(English Series)(2020年1期)2020-04-27 08:15:42
质量安全与检验检测(2019年2期)2019-05-18 05:39:24
小星星·阅读100分(低年级)(2018年4期)2018-05-07 07:25:40
小猕猴智力画刊(2017年9期)2017-10-19 01:01:57
新高考(英语进阶)(2016年11期)2017-01-15 14:22:34
学苑创造·A版(2016年1期)2016-03-10 18:17:18
新作文(小学中高年级版)(2015年5期)2015-04-12 06:27:42
中国医院用药评价与分析(2015年1期)2015-01-08 03:54:10
