
S1核酸酶介导的功能核酸生物传感器研究进展
2019-01-23 12:05张媛田晶晶罗云波田洪涛许文涛
生物技术通报 2019年1期
张媛 田晶晶 罗云波 田洪涛 许文涛
(1. 河北农业大学食品科技学院,保定 071001;2. 中国农业大学 北京食品营养与人类健康高精尖创新中心,北京 100083;3. 农业部农业转基因生物安全评价(食用)重点实验室,中国农业大学,北京 100083;4. 国家北方山区农业工程技术研究中心,保定 071001)
生物传感器是以具有生物识别作用的生物成分(如酶、蛋白质、DNA、抗体和抗原等)或生物体本身(如细胞、微生物和组织等)为敏感材料,与适当的信号转换器相结合,将生化反应的程度用离散或连续的信号表达出来,从而能够进行生命物质和化学物质检测和监控的装置[1]。近年来,生物传感器已经得到了空前的发展,并且已经应用于食品、医药、环境等各个领域,生物传感器的种类有很多,其中核酸生物传感已经成为近几年大量学者的研究热点。
核酸生物传感器是利用DNA分子之间互补配对的特异性,以核酸作为分子识别元件,将反应产物转变为电、光、声等可识别的信号,实现对特定核酸序列进行分析的传感器。由于其具有简便快速、灵敏度较高、稳定性强及成本较低的特点,广泛应用于临床医学诊断、食品检验、环境监测等领域。各种核酸内切酶在核酸生物传感器的构建中都起了重要作用,如双链特异性核酸酶(Duplex-specific nuclease,DSN酶),根据其对单双链核酸不同的切割特点,搭载不同的信号输出及扩增方式,已经介导了一系列核酸传感器包括光学传感器、电化学传感器和光磁传感器等,实现了microRNA、mRNA、重金属离子等一系列生物标志物及食品安全风险因子的检测。脱嘌呤/脱嘧啶核酸内切酶1(Apurinic/apyrimidinic endonuclease 1,APE1),具有能够在碱基缺失位点(AP site)处识别并切割 DNA 能力,利用这一能力可以生成理想的功能核酸链,并结合不同的信号输出及放大方式,研究者已经建立了一些 APE1介导的电化学、荧光功能核酸生物传感技术,实现了对 DNA 糖基化酶等的酶活性的检测,同时也对肿瘤的治疗方面有一定的应用。末端转移酶(Terminal deoxynucleotidyl transferase,TdT酶)是一种无需模板的DNA聚合酶,可以催化DNA的3′末端添加同聚物或在DNA的3′末端标记,可以介导电化学、荧光等核酸传感器,用于检测金属离子,蛋白质分子和小分子等,也能诊断、治疗如急性淋巴性白血病等疾病。
S1核酸酶是一种高度单链特异的核酸内切酶,来源于稻谷曲霉(Aspergillus oryzae),是由Vogt在1973年首先从米曲霉中分离得到的[2],广泛用于核酸的生物化学分析。目前,结合S1核酸酶对核酸链特有的性质及生物传感器快速、微量检测的优点,人们设计出了许多精确度越来越高的S1核酸酶介导的核酸生物传感器,可以用来检测不同种类的金属离子、单链核酸、氨基酸等,S1核酸酶的应用也越来越广泛。因此本文对近年来S1核酸酶的应用,介导的生物传感器的种类等进行了总结,有助于使其在生物学、分子诊断学、基因组研究和药物开发等领域得到更好的应用。
1 S1核酸酶的介绍
1.1 S1核酸酶的结构和功能
S1核酸酶是S1-P1核酸酶家族的一种锌离子依赖性核酸酶,是具有两个N-糖基化位点的糖蛋白,S1-P1核酸酶家族在真菌、植物、原生动物和寄生虫等中广泛存在,但S1核酸酶只存在于稻谷曲霉中。Kovaľ等[3]展示了这种核酸酶的第一个X射线结构,以及对负责识别和结合配体的反应、抑制机制、性质进行了彻底分析,如鉴定出了额外的核碱基结合位点,明确了活性位点与磷酸盐、核苷酸等的多种结合方式。S1核酸酶能降解单链DNA或RNA,对双链DNA、双链RNA和DNA-RNA杂交体相对不敏感。通常水解单链DNA的速率要比水解双链RNA快75 000倍。这种水解反应在细胞内的生物过程以及许多生物技术中发挥着重要的作用,如基因的修复、重组、复制、转录以及分子克隆、基因分型和映射等[4-8]。
1.2 S1核酸酶的性质
S1核酸酶是一种分子量为32 kD,依赖 Zn2+的糖蛋白质,最适pH范围为4.0-4.3。该酶对热很稳定,在底物存在下65-70℃仍能表现活性。经研究表明,一些螯合剂(如EDTA和柠檬酸等)能强烈地抑制S1核酸酶活性,K+以某些G-四联体为底物对S1核酸酶的活性有抑制作用[9],0.4 mmol/L的Na2ATP可以有效的抑制S1核酸酶对ssDNA的消化[10]。此外,磷酸缓冲液也可以抑制其活性,但它对尿素及甲酰胺等试剂是稳定的。
S1核酸酶尽管其主要底物是单链的,但它也可以偶尔在双链DNA、RNA或DNA-RNA杂合体中引入单链断裂。即它可以水解双链DNA中的单链区域,从单链部位切断核酸分子,而且这种单链区可以小到只有一个碱基对的程度,如环和间隙。由于具备这种功能,使S1核酸酶在分析核酸杂交分子(RNADNA)的结构、RNA分子定位、测定真核基因中间隔子序列的位置、去除DNA片段中突出的单链尾,以及打开在双链cDNA合成期间形成的发夹环过程中起作用。更重要的是,S1核酸酶不需要特定的识别位点和 3′-5′(或 5′-3′)方向的水解,因此无论发夹的末端或茎环的序列如何,都会发生水解作用。这与早期分子信标,纳米粒子扩增方案或量子点耦合荧光共振能量转移中使用的核酸内切酶形成鲜明对比[11-15]。
2 S1核酸酶介导的功能核酸生物传感器分类
2.1 S1核酸酶介导的肽核酸(PNA)生物传感器
肽核酸(Peptide nucleic acid,PNA)是具有类多肽骨架的DNA类似物,可以特异性地与DNA或RNA杂交,形成稳定的复合体。Ye等[16]通过结合PNA和S1核酸酶开发了一种非常简单和实用的基因分型方法。样品DNA中与PNA互补(完全或部分)的部分被保护,可免于酶的消化,通过添加识别PNA / DNA双链体的3,3′-二乙基硫代二羰花青碘化物可以通过比色法判断是否有SNP存在,这种化合物在PNA与DNA完全互补时混合物呈紫色,有SNP存在时呈蓝色。可以将这种方法应用于医学领域,并用肉眼观察颜色的变化来快速检测SNP是否存在于患者的DNA中。
2.2 S1核酸酶介导的表面增强拉曼散射(SERS)
传感器
Yoo等[17]将金纳米线膜上表面增强拉曼散射(Surface enhanced raman scattering, SERS)平台与 S1核酸酶反应相结合,开发了一种新型的用来检测错配碱基(Single nucleotide polymorphisms,SNPs)的传感器。将探针DNA通过巯基连接到金纳米线(Au nanowire,Au NW)上,当目标DNA浸入含有探针DNA的溶液中时,探针DNA可以与目标DNA杂交。如果目标DNA与探针DNA杂交完全匹配,则其保护探针DNA免受S1核酸酶水解,使得拉曼信号分子Cy5保留在Au NW附近的探针DNA处,由此提供强烈的SERS信号。如果与探针DNA杂交的靶DNA具有错配,则通过S1核酸酶的水解除去Cy5并且拉曼信号消失。从用S1核酸酶处理之前和之后的拉曼信号的差异可以清楚地识别出SNPs的存在。
Draz等[18]研发了一种将环介导等温扩增(Loop mediated isothermal amplification,LAMP)和表面增强拉曼散射相结合的DNA感应测定法,应用的金纳米探针专门设计用于标记LAMP产生的DNA扩增产物,使用SERS技术进行检测,原理如图1所示。依靠使用S1核酸酶的酶消化步骤来区分靶DNA的存在与否,当靶DNA存在时可特异性结合其互补序列并形成双链DNA结构,其中复杂的cy5-DNA被保护免受核酸酶消化,并且最终cy5保持与金纳米颗粒的表面连接,产生较强的拉曼信号。相反,在没有靶DNA的情况下,S1核酸酶通过水解金纳米探针的表面的DNA寡核苷酸,触发从金纳米颗粒表面释放cy5,导致显著降低拉曼信号的强度。
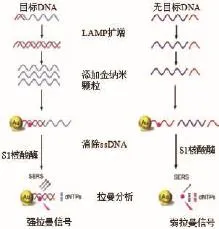
图1 LAMP-SERS检测实验原理图[18]
2.3 S1核酸酶介导的荧光传感器
Guo等[19]开发了一种新型的无标记荧光生物传感器,实验基于阳离子(9,9-双(6′-N,N,N-三甲基铵)己基)氟亚苯基(9,9-bis(6′-N,N,N-trimethylammonium)hexyl)fluorine phenylene,PFP)和苝二酰亚胺衍生物(Perylene diimide derivatives,PDI)开发了一种新型的无标记的荧光生物传感器。由于PFP和PDI两者之间具有静电排斥作用,PDI本身不能淬灭PFP的荧光。当ssDNA被添加到混合溶液中时,由于更强的静电吸引相互作用,可以形成PFP / ssDNA / PDI复合物,使荧光淬灭。当存在S1核酸酶时,ssDNA被水解成小片段,PFP / ssDNA / PDI复合物被分解并且小ssDNA片段上的PDI无法聚集,PDI的淬灭能力减弱与PDI之间的距离较远,使PFP的荧光显着恢复。该方法更灵敏、高效及简单,为生物分析提供了一个新的平台。
Bao等[20]采用阳离子共轭聚电解质(Conjugated polyelectrolytes,CPEs)作为检测DNA的指标,开发了新型均相杂交链式反应(Hybridization chain reaction,HCR)检测方法。实验采用两个用荧光素标记发夹探针H1和H2,在存在目标ssDNA时,经过HCR之后,两个发夹探针被靶标DNA触发以形成在链中具有多个荧光素标记的长链双链DNA聚合物,其对S1核酸酶降解具有抗性。当添加CPE时,荧光素标记的DNA与CPE之间发生强烈的静电相互作用,从而允许从CPE到荧光素标记的DNA的荧光共振能量转移(Fluorescence resonance energy transfer,FRET)以及共轭聚合物溶液的明显的荧光颜色变化,荧光颜色由蓝色变为绿色。在不存在目标ssDNA时,S1核酸酶可以结合并裂解H1和H2发夹探针的单链DNA片段,这导致H1和H2的茎环区释放荧光素。在UV照射下可以观察到阳离子共轭聚电解质[(9,9-双 {60 -[(N,N-二乙基)-N-甲基铵]己基} -2,7-亚麻烯基乙炔)-共 -(2,5-双 {30 -[(N,N-二乙基)-N-甲基铵]-10-氧杂丙基 } -1,4-亚苯基)]四碘化物([(9,9-bis{60-[(N,N-diethyl)-N-methylammonium]hexyl}- 2,7- fuorenyleneethynylene)-alt-co-(2,5-bis{30-[(N,N-diethyl)-N-methylammonium]-10-oxapropyl}-1,4-phenylene)]tetraiodide,PFEPN+)发出的蓝色荧光,原理图如图2所示。
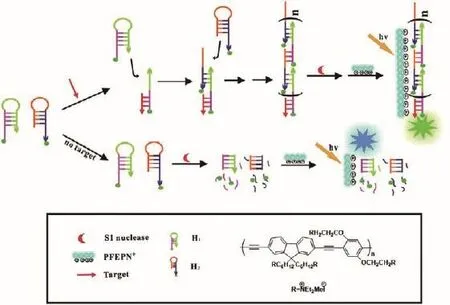
图2 基于HCR和CPE的DNA检测级联信号放大策略示意图[20]
2.4 S1核酸酶介导的纳米材料传感器
贵金属纳米簇(Nano-cluster,NCs)因其尺寸小、无毒性和生物相容性良好等优点而引起了广泛关注。银纳米簇(Ag nanocluster,AgNCs)作为一种贵金属纳米簇,因具有高度极化的电子跃迁性能,已成为一种极具潜力的荧光基团[21-22]。Wang等[23]以富含胞嘧啶C的单链DNA作为模板合成高荧光量子产物AgNCs,当S1核酸酶存在时,S1核酸酶可把ssDNA降解为单核苷酸或寡核苷酸片段,使其无法进行构型转变形成AgNCs,导致体系荧光信号显著降低。该荧光探针方法灵敏度高、操作简单且无需标记。
Qi等[24]基于(+)金纳米粒子(Au nanoparticles,AuNPs)对长ssDNA和由酶解切割反应产生的片段化DNA的完全不同的电化学发光(Chemiluminescence,Cl)效应,来监测核酸酶对DNA切割过程。鲁米诺阴离子和氢过氧化物阴离子(HO2-)是Cl系统中存在的主要分子形式,ssDNA本身是带有负电荷磷酸骨架的聚阴离子物质,通过静电相互作用与(+)AuNPs作用,导致Cl反应体系的局部浓度降低,使得化学发光强度降低。当S1核酸酶存在时,ssDNA被S1核酸酶切割成小片段,一方面,片段化的DNA具有较少的带负电荷的磷酸骨架其电负性较弱;另一方面,小DNA片段不能像长的ssDNA那样自由地包裹AuNPs表面,因此片段化DNA不能与(+)AuNPs相互作用,(+)AuNPs保持其原始的高正电荷密度,并且增加的局部浓度效应产生强Cl信号,原理图如图3所示。
3 S1核酸酶介导的功能核酸生物传感器的应用
3.1 金属离子的检测
钾离子(K+)是细胞内液中主要的阳离子,心肌和神经肌肉均需要相对恒定的钾离子浓度以维持正常的应激性,同样也是植物生长过程中所需要的重要营养物质[25]。精确的检测K+这类物质,在生物化学、临床医学及食品卫生等方面有着重要的意义。Zhou等[9]利用目前研究的以原卟啉IX(Protoporphyrin IX,PPIX)作为信号源,开发的基于G四联体的内切核酸酶活性荧光的测定法进行研究,当S1核酸酶能够将G-四联体DNA切割成小片段即不会形成G-四联体时,PPIX的荧光会减弱。在加入K+后能降低S1核酸酶的活性,稳定G4结构,使PPIX的荧光可以大大提高,该系统可以选择性的检测K+。
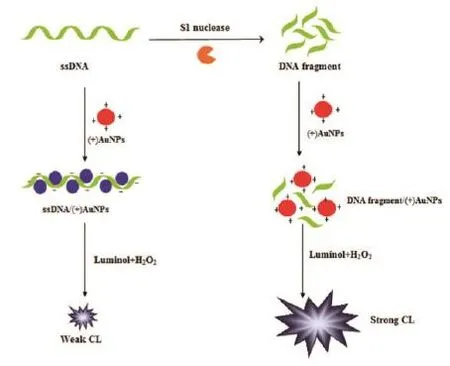
图3 通过Cl信号来监测核酸酶对DNA切割过程原理图[24]
Pb2+是生态环境中毒性最大的污染物之一,会对人体健康,特别是儿童健康产生严重影响。找到一种方法简单,操作简便,性价比高,并且具有良好的灵敏度和选择性的检测方法尤为重要。Li等[10]用凝血酶结合适体(Thrombin adaptor,TBA)作为酶结合适体,在没有Pb2+的情况下,核酸酶S1可以将单链TBA有效降解为单核苷酸或寡核苷酸片段,因此TBA片段不能诱导探针的聚集形成,导致弱的荧光强度。在Pb2+存在下,TBA折叠成稳定的G4结构,其阻断核酸酶S1的水解消化,聚集复合体预计会形成,荧光强度会增强,原理如图4所示,实验发现荧光发射的强度与Pb2+的添加量成正比。
3.2 单链核酸的检测
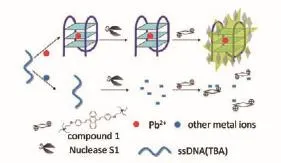
图4 检测pb2+的原理示意图[10]
Micro RNA是一类短小的、内源性的非编码RNA,是单链核酸的一种,长度大约在18-24个核糖核苷酸之间。它们在生理和病理过程中扮演重要的角色,可以调控基因的活性,且对细胞的增殖、迁移以及凋亡有抑制或促进作用[26-27],对癌症、糖尿病和阿尔兹海默症的诊断也十分有价值[28]。
Micro RNA被当作癌症治疗中的肿瘤标记物和治疗靶标[26-27,29-30]。因此,micro RNA的高效检测对更好地了解它们在肿瘤细胞中的角色,以及进一步验证其在生物医学研究和临床诊断中的功能至关重要。但是,micro RNA的一些特殊性质使分析它们具有一定的难度。例如,片段小、在家族中序列的高同源性、在总体RNA中的低丰度等[31]。因此,发展特异性、超灵敏的micro RNA定量检测的方法乃当务之急。Zhou等[32],利用S1核酸酶只能水解单链核苷酸不能水解双链DNA、RNA或DNA-RNA杂合体这一特殊的性质,开发出一种简单的电化学方法用于micro RNA的检测,在不存在杂交过程的情况下,电极表面上的组装探针DNA可以被核酸酶S1容易地消化,并且由于朝向氧化还原探针可以降低排斥力产生的强电化学信号。然而,当探针DNA与靶micro RNA杂交后,核酸酶S1的消化活性被抑制,这可能导致弱的电化学信号。基于电化学信号的变化,可以实现靶microRNA-319a的检测,实验原理如图5所示。
3.3 小分子的检测
腺苷三磷酸(Adenosine triphosphate,ATP)被认为是生命的能量分子,作为细胞内能量传递的“分子货币”,起着储存和传递化学能的作用,其参与着机体的新陈代谢[33],在生物合成,神经活动,主动运输等方面也发挥着重要的作用。传统方法如高效液相色谱法,虽然能检测出ATP,但通常耗资高,方法复杂,实验操作繁琐。Liu等[33]基于滚环扩增(Rolling circle amplification,RCA)和Exo III辅助循环的双重放大荧光传感平台,利用ATP对S1核酸酶活性的抑制作用,从而保护RCA反应的引物链不被水解,使RCA反应得以进行,并顺利地发生后续的核酸外切酶III辅助荧光信号放大反应,实现对ATP的检测。
3.4 氨基酸的检测
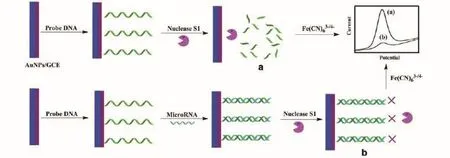
图5 基于针对核酸酶S1消化的杂交保护的micro RNA测定的检测策略的示意图[32]
2011年,Xu等[34]利用S1核酸酶在单链DNA上的水解功能,提出了一种灵敏的检测L-精氨酰胺的方法。用可以产生化学发光信号(Cl)的试剂N-(4-氨基丁基)-N-乙基异鲁米诺[N-(4-aminobutyl)-N-ethylisoluminol,ABEI]标记DNA适体及其互补DNA。将用5′-ABEI标记的DNA1-ABEI固定在胶体金覆盖的磁珠(MB)上,DNA1为L-精氨酸酰胺适体互补序列,用3′-ABEI标记的DNA2-ABEI以MB双链形式自我装配,DNA2为L-精氨酸酰胺适体。在目标L-精氨酰胺的存在下,形成茎环适体结构,其响应地使双链体变性并从互补DNA2释放,形成DNA2中的单链区域。S1核酸酶催化逐步去除单链区域的单核苷酸并最终释放L-精氨酰胺。释放的L-精氨酰胺然后与另一个适体相互作用,从那里重新开始循环。同时,MB表面形成的单链DNA也被S1核酸酶水解。因此,单一的L-精氨酰胺产生许多Cl试剂ABEI,进而产生强烈的Cl信号。实验原理如图6所示。
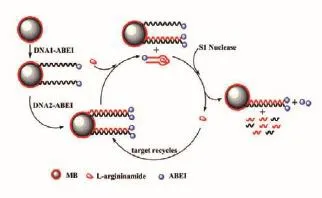
图6 灵敏的检测L-精氨酰胺的原理图[34]
3.5 核酸反应体系的纯化
Yoo等[35]开发了基于S1核酸酶的多点阵列系统,该系统可高通量地检测出错配DNA,有效防止出现假阴性结果,具有高度特异性。在杂交期间,靶标DNA结合到探针DNA的匹配区域形成DNA双链体(dsDNA),没有互补序列区的和不能与任何靶标DNA结合的探针DNA 或者形成具有由一个或多个错配碱基对产生的单链环的异源双链体可被S1核酸酶水解,将完美匹配的探针DNA留在玻璃基底上,在玻璃基底可以检测到荧光信号。这种方法也可以用来检测导致传染性疾病的基因突变,SNPs,遗传疾病和微生物病原体等。
Guan等[36]设计了一种检测复杂的污染物样品中的序列特异性单链DNA(ssDNA)简单的无标记传感方法,检测原理如图7所示。在检测过程中,S1核酸酶起着重要的作用。S1核酸酶可以降解单链核酸或在由切口,缺口,错配或环引起的单链区裂解双链DNA,核酸染料只能与dsDNA结合,通过使用S1核酸酶清除多余的互补ssDNA和可能错配的双链DNA,消除过多的互补单链DNA或其他单链核酸污染物,从而降低非特异性荧光背景,进而提高所提方法的灵敏度和实用性。
3.6 多元化基因文库的构建
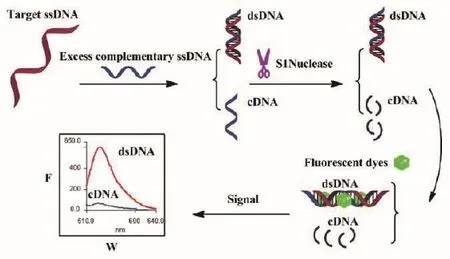
图7 检测ssDNA的原理图[36]
将含有某种生物不同基因的许多DNA片段,导入受体菌的群体中储存,各个受体菌分别含有这种生物的不同的基因,称为基因文库。Tullman[37]研究发现S1核酸酶可以通过在质粒分子中产生单一的双链断裂将超螺旋质粒DNA转化为单位长度的线性双链DNA,这些双链断裂不仅发生在靠近反向重复的复制起点处,而且遍及整个质粒的各个位置。S1核酸酶在通常用于单链核酸酶活性的条件下才显示出这种活性。因此,质粒DNA的S1核酸酶消化在第一次双链断裂后有效停止。该属性使构建大型域插入库变得更加容易,其目标是在各种位置插入线性DNA。所以,利用S1核酸酶这一性质能够构建更多元化的基因文库。
4 S1核酸酶的检测
传统的检测S1核酸酶的方法,如放射性同位素标记法、聚丙烯酰胺凝胶电泳法、高效液相色谱法及酶联免疫分析法等。这些方法费力、耗时而且操作较为复杂,部分方法还存在需要同位素标记的缺点。近些年来,为了克服这些缺点,其它检测核酸酶活性的技术相继出现,如电化学技术[38]和光学技术。例如,一些可附着在DNA上的分子,如硅杂环戊二烯分子、二萘嵌苯衍生物分子[39]及金纳米颗粒[40],一旦核酶将DNA水解,将导致这些分子的聚集状态发生改变,进而引起溶液的荧光或颜色改变,以此来检测核酸酶。
He等[41]设计了一种基于氧化石墨烯(Graphene oxide,GO)的传感系统用于核酸内切酶的检测。研究表明,GO有淬灭荧光的作用并且可以与单链DNA相互作用,实验采用20-mer ssDNA作为核酸酶底物,20F通过p-p堆积被吸附到GO片上,由此GO显着地猝灭荧光基团-羧基荧光素(Financial analyst meeting,FAM)的荧光。在S1核酸酶的存在下,可将20F切割成片段,由于短的FAM连接的寡核苷酸片段对GO的弱亲和力,将GO引入感应溶液导致FAM荧光弱猝灭,并且随着S1核酸酶浓度的增加,荧光强度逐渐增强,FAM的荧光强度与S1核酸酶的浓度成正比。
Li等[42]将阳离子 9,9-双(6,6-(N,N,N-三甲基铵)-芴)-2,7-亚基-亚乙烯基-共-交替-2,5-二氰基 -1,4-亚 苯基(9,9-bis(6,6-(N,N,N-trimethylammonium)-fluorene)-2,7-ylenevinylene -co -alt-2,5-dicyano-1,4-phenylene),PFVCN) 用 作 信 号 物质,单层二硫化钨(Tungsten disulfide,WS2)可作为带负电荷表面的猝灭剂。实验开始时,PFVCN与WS2反应使荧光淬灭,当ssDNA存在时,由于更强的静电作用,ssDNA与PFVCN相互作用形成复合物,PFVCN发出黄色荧光。当ssDNA被S1核酸酶或羟基自由基水解成小片段时,片段化的DNA和PFVCN之间的相互作用变弱,导致PFVCN被吸附在WS2的表面上并且通过荧光共振能量转移淬灭荧光。
5 总结与展望
综上所述,由于S1核酸酶特有只能水解单链DNA和RNA而不能水解双链DNA和RNA的这一性质,其已广泛的被应用于生物传感器的检测领域。等温核酸扩增技术是一类新型的核酸分析技术,实现了恒温条件下核酸的扩增。与传统PCR相比,等温核酸扩增技术摆脱了对热循环仪器的需求,并且针对靶标序列的扩增具有耗时短、反应灵敏、特异性高等优点,越来越多的科研人员也开始把目光聚焦于体外核酸扩增新技术的开发。S1核酸酶室温下就可发生反应,且具有较高的热稳定性,因此可与体外恒温核酸扩增技术如LAMP、RCA等相结合实现信号放大并实现靶物质的检测。目前S1核酸酶介导的生物传感器可用于检测金属离子、micro RNA、小分子物质、氨基酸等,由于其可降解错配碱基,也可用于纯化反应体系,人们设计出了许多精确度越来越高的生物传感器用来检测S1核酸酶,但总体来说S1核酸酶的应用较少,如何利用S1核酸酶设计高灵敏度、高特异性的生物传感器将成为今后研究的热点问题。
总体来说,对S1核酸酶的研究仍处于初级阶段,要想使其介导的生物传感器得到更广泛的应用,需要在以下几方面进行进一步研究和探索:首先,在理论方面需要深入研究S1核酸酶的结构与活性的构效关系,找出其活性位点,探究其对于核酸链的碱基碎片化规律,为其应用提供理论支持。其次,应寻找一种方法可以使该酶的活性被封闭,便于常温贮藏和运输,使其能在快速检测领域发挥作用。再次,可利用该酶与其他酶,如解旋酶、逆转录酶等,配合开发新的复合酶恒温扩增新技术,并探究复合酶的最佳应用场景及最优反应体系。该酶核酸酶介导的生物传感器主要为荧光生物传感器,对于其他类型的生物传感器涉及的较少,并且一般都为单一物质检测,我们应该合理的利用S1核酸酶开发出功能多样化、智能化集成化、微型化的生物传感器。最后,该酶还可与新型纳米材料结合运输到胞内及体内进行活细胞生物传感器的检测,基于S1核酸酶的胞内检测和胞内成像将成为未来的研究热点。
猜你喜欢
海外星云 (2021年21期)2021-01-19
无机化学学报(2020年7期)2020-07-20
中国预防兽医学报(2020年2期)2020-06-01
三农资讯半月报(2020年8期)2020-05-13
中国海洋大学学报(自然科学版)(2019年2期)2019-12-07
天水师范学院学报(2018年5期)2018-12-04
智富时代(2018年9期)2018-10-19
智富时代(2018年9期)2018-10-19
中国病理生理杂志(2017年2期)2017-01-17
当代经济(2016年26期)2016-06-15
