
一种新型铁电移相器材料的第一性原理计算
2019-01-17 01:07张良俊侍述海吕宇宙
上海航天 2018年6期
张 杨,张良俊,侍述海,姜 伟,叶 舟,吕宇宙
(上海航天电子通讯设备研究所,上海 201109)
0 引言
铋层状结构铁电(BLSFs)薄膜在用于相控阵雷达的铁电移相器中具有极大应用价值,受到广泛的关注。移相器的技术特征要求移相器材料本身具备较大的自发极化和良好的抗疲劳属性[1-2]。因此,在这些材料中,SrBi2Ta2O9(SBT)和Bi4-xLaxTi3O12(BLT)被认为是最有可能替代传统PbZr1-xTixO3(PZT)基钙钛矿铁电的铁电移相器材料[2-7]。国内也有相关报道表明,在SAR成像中铁电材料移相器是未来的发展趋势[8-9]。尽管SBT和BLT都具有优异的抗疲劳特征(SBT的极化翻转次数可超过1012),但相对较高的处理温度和较低的剩余极化制约了其在实际中的应用[1,10-12],而离子掺杂取代被认为是解决此类问题的主要途径。
SrBi2Nb2O9(SBN)是另外一类具有跟SBT类似的,含有铌铋层状结构的铁电体[13]。与SBT相比,SBN同样具有优异的抗疲劳属性,且SBN有着更高的居里温度(SBN,440 ℃;SBT,335 ℃)[14]。通过中子和电子衍射实验发现,SBN中NbO6的畸变程度要大于SBT中TaO6的畸变程度,这有利于SBN产生更大的自发极化,说明SBN也是一种非常有潜力的铁电移相器侯选材料。实验发现,如果SBN中部分A位阳离子(Sr2+)被Ba2+取代,也会出现与BaBi2Nb2O9(BBN)类似的介电驰豫行为,极大地丰富了SBN的应用范围[15]。同时,实验发现,用其他金属阳离子(如Bi3+, Pb2+, Ca2+等)部分取代SBN中A位阳离子(Sr2+),SBN的居里温度、极化强度及其介电属性能会得到极大改善[16]。因此,独立研究不同的A位阳离子在铋铌酸盐ABi2Nb2O9(A=Ba, Pb, Sr, Ca)体系中的电子结构和微观属性,对理解ABi2Nb2O9特定的铁电性质和构造出新的铁电移相器材料具有重要意义。
块体铋铌酸盐ABi2Nb2O9铁电体(A=Ba, Pb, Sr, Ca)具有跟SBT类似的晶体结构,如图1所示。ABi2Nb2O9晶体结构起源于一个非极化高温四方结构(空间群为I4/mmm),晶胞内包含一个原胞(见图1(a)),室温下正交结构(空间群为A21am)包含2个原胞(见图1(b))。
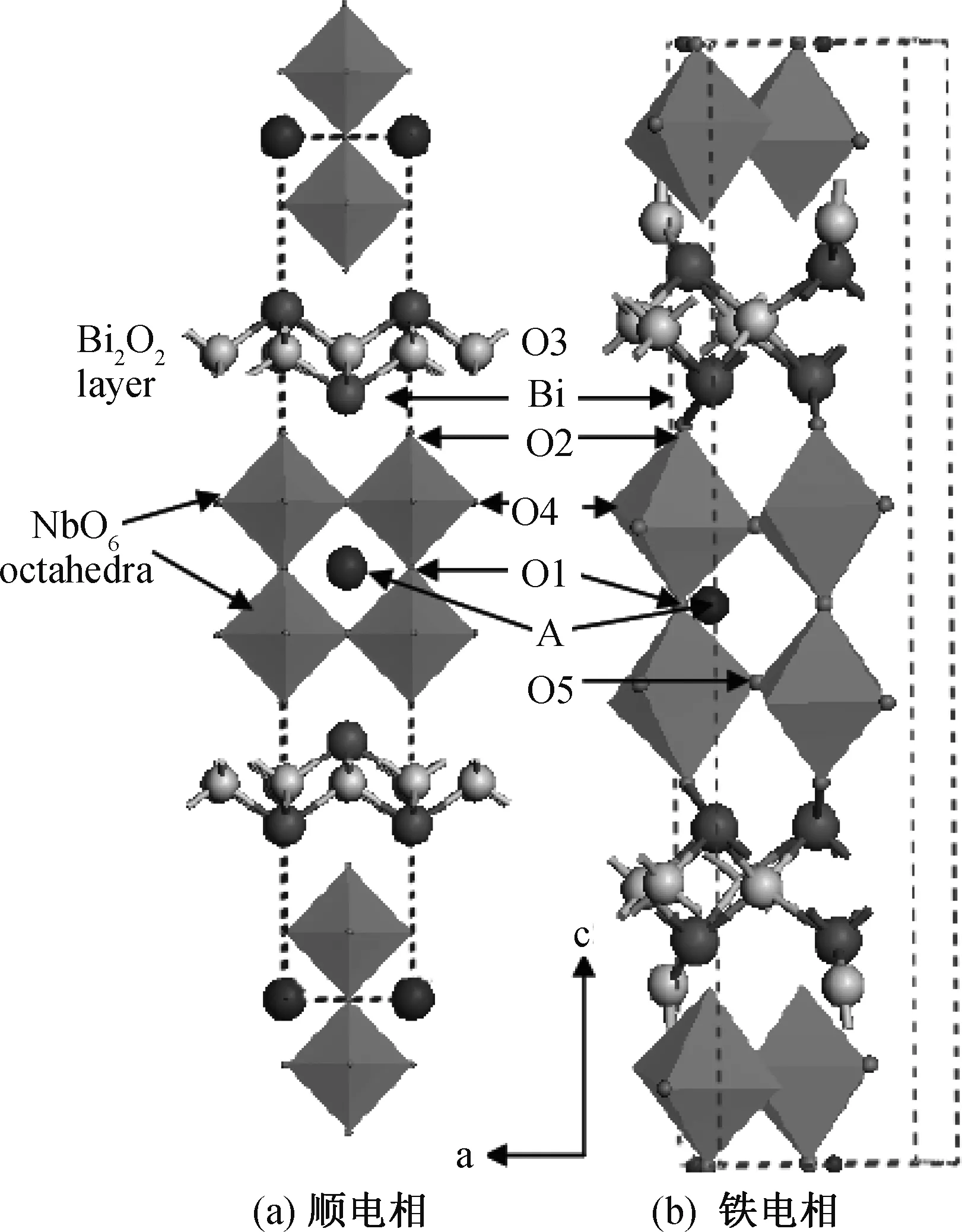
图1 ABi2Nb2O9(A=Ba, Pb, Sr, Ca)铁电体在顺电相和铁电相中的晶体结构Fig.1 Crystal structures of ABi2Nb2O9 (A=Ba, Pb, Sr, Ca) ferroelectrics in paraelectric phase and ferroelectric phase
本文采用第一性原理密度泛函理论(density functional theory,DFT)综合研究ABi2Nb2O9(A=Ba,Pb,Sr,Ca)体系结构、电子性质、化学键及自发极化,阐述了ABi2Nb2O9铁电体的铁电起源,研究了这些材料电子结构和极化属性随A位离子变化趋势,并运用现代极化理论计算了SBN铁电极化,同时对BBN,PBN,CBN的极化强度给出理论预测。
1 第一性原理计算方法
本工作中的计算基于第一性原理密度泛函理论,使用缀加投影平面波(PAW)的方法,计算实施通过第一性原理计算软件包(VASP)的软件包,交换关联能使用广义梯度近似(GGA)来描述。
化学键的分析涉及在成键过程中电荷迁移的问题,BADER的AIM理论为准确定义一个原子内电荷量提供了有力的支持。在这种方法里,一个原子被定义为包含单个原子核,并由电荷密度梯度为零的表面所组成的空间,即
ρ(r)·n(r)=0
(1)

2 结果和讨论
2.1 体系的基态结构

(2)
NbO6八面体单元和Bi2O2层的结构畸变参数Δ1和Δ2见表1。结果表明:在铁电相变的过程中,NbO6八面体的畸变程度远大于Bi2O2层的畸变,这说明自发极化主要来源于NbO6八面体内离子的位移。A位离子对NbO6八面体的作用,使不同体系中的NbO6的畸变参数有所差异,集中体现在BaBi2Nb2O9的NbO6畸变参数小于其他ABi2Nb2O9(A=Pb, Sr, Ca)体系的畸变参数,这表明:BaBi2Nb2O9中的A位离子(Ba2+)半径占据了Nb-O6畸变空间,使得在相变时Nb-O键长的活跃范围变小。而PbBi2Nb2O9相对较大的畸变参数来源于A位离子(Pb2+) 的6s态对整个系统的电子态的影响,Pb2+6s态的作用将在下面部分进行具体讨论。
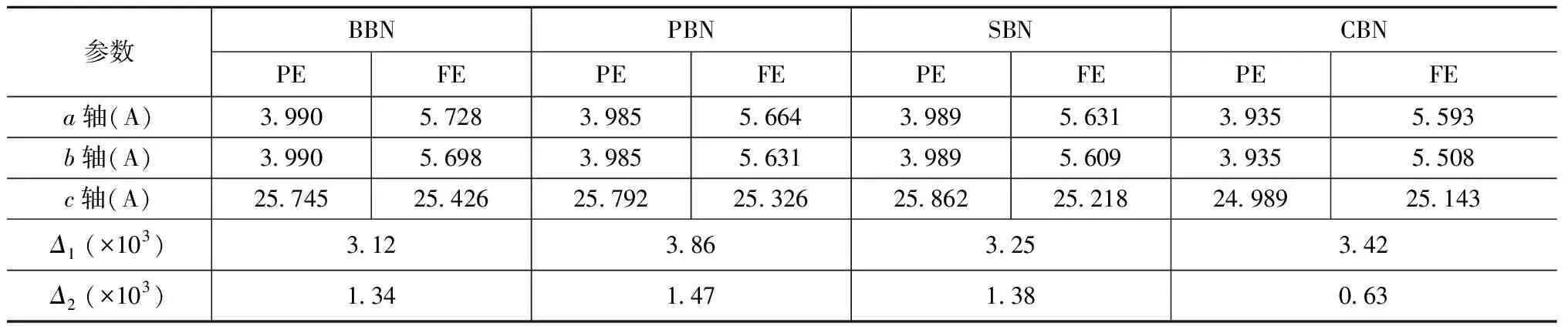
表1 ABi2Nb2O9的晶格常数及NbO6八面体和Bi2O2层的畸变参数Tab.1 Lattice constant of ABi2Nb2O9 and distortion parameters of NbO6 octahedron and Bi2O2 layer
2.2 电子结构和铁电属性
ABi2Nb2O9(A=Ba,Pb,Sr,Ca)总态密度如图2所示,其中实线和点线分别代表的是铁电相和顺电相,系统显示了绝缘特征。ABi2Nb2O9的顺电相和铁电相对应的带隙如图3所示。显然,铁电的相变导致了带隙显著的增加(≥0.75 eV)。A位离子的大小强烈影响着体系的带隙,顺电相时ABi2Nb2O9带隙随着A位离子从Ba2+变化到Pb2+,Sr2+,Ca2+(A=Ba2+→Ca2+)有大约0.1 eV的增加,铁电相时,对应着A=Ba2+→Ca2+,带隙从2.10 eV增加到2.67 eV。在铁电相时带隙出现明显的变化一定程度上反映了A位离子在相变过程中影响着ABi2Nb2O9电子结构的变化,带隙的增加降低了材料本身的漏电流,有利于移相器件的密集化。图2中,从态密度的贡献上来看,价带主要是由O2-的2p态构成,也包含部分的Bi3+的6s和6p态及Nb5+的4d态的贡献,导带除了Nb5+的4d和Bi3+的6p态的贡献外,还包含了一部分的O2-的2p态的贡献。对于BBN和PBN来说,价带还包括明显的Ba2+的5p态和Pb2+的6s态的贡献。轨道之间的交叠说明Nb-O和Bi-O之间存在强烈的轨道杂化。

图2 基态ABi2Nb2O9 (A=Ba,Pb,Sr,Ca)铁电相总态密度及阳离子的部分态密度Fig.2 Total ferroelectric phase state density of ground state ABi2Nb2O9 (A=Ba, Pb, Sr, Ca)

图3 ABi2Nb2O9 (A=Ba,Pb,Sr,Ca) 顺电相和铁电相对应的带隙Fig.3 Gaps of ferroelectric phase and paraelectric phase of ABi2Nb2O9 (A=Ba, Pb, Sr, Ca)
O2-的2p态,Nb5+的4d态,Bi3+的6s态和Bi3+的6p态是ABi2Nb2O9体系价带和导带的主要贡献,因此Nb-O和Bi-O之间强烈的轨道杂化也是ABi2Nb2O9体系铁电畸变的主导因素。由于铋铌酸盐体系具有相似的Nb5+,Bi3+和O2-的电子能态变化,因此这里只针对SBN的顺电相和铁电相的态密度进行分析。通过分析,可以发现铁电畸变带来3个显著的变化:1) Nb5+和Bi3+的贡献在导带部分往高能端移动造成带隙的增加;2)Nb5+和Bi3+的贡献在价带的高能端部分下降,而在低能端部分上升;3)顺电相时以-9.8 eV为中心的Bi3+的6s态在铁电相时处于-8.7 eV。Nb5+和Bi3+的能态在价态部分的变化使Nb-O和Bi-O之间形成更强的杂化,降低了原子间的短程排斥力,有利于铁电的畸变Bi3+的6s态在铁电相变过程中向高能方向上的移动,说明单纯的Bi-O之间的杂化并不能导致铁电相的晶格失稳。在这一点上,Bi3+的行为并不同于Pb2+在PbTiO3中的作用,在PbTiO3中,Pb2+的6s态在相变过程中向低能方向移动,直接降低了总能,从而有利于铁电相变。尽管Bi-O间的杂化不能直接导致铁电相变的稳定,但是Bi-O间的杂化使得部分电荷从O2-转移到Nb5+和Bi3+,有利于进一步加强Nb-O之间的杂化,以达到降低晶体总能和稳定铁电相的目的。
此外,图2中基态ABi2Nb2O9铁电相总态密度也体现出一定的差异,这种差异主要是来源于A位离子对ABi2Nb2O9铁电相总态密度的影响。为了解释A位离子的作用, 对A位离子的分态密度进行计算,如图4所示。

图4 ABi2Nb2O9系统的A位离子的分态密度Fig.4 Partial densities of states of A-site ions in ABi2Nb2O9 system
从对价态的贡献来看,活跃的Pb孤立电子对价态的贡献远大于其他A位离子。另一方面,离子半径的大小也影响着A位离子对态密度的贡献,可以发现随着离子半径的减小(从Ba2+到Ca2+),A位离子在价带态密度的贡献也随之减小。A位离子在价带部分的贡献使A-O之间发生杂化,这说明A位离子与相邻的氧离子存在共价作用,而这种共价作用必然引起价态电荷的迁移,从而影响到其他原子的电子能态变化,由图5中基态ABi2Nb2O9铁电相的Nb和Bi原子分态密度可发现,PBN对应的Nb的4d态,Bi的6s态和6p态的态密度稍微移向低能端,而CBN对应的分态密度稍微移向高能部分。不同的ABi2Nb2O9的Nb5+和Bi3+分态密度的分布反映了A位的阳离子与O2-的2p之间的杂化程度。杂化越强,共价性质越强,从O2-返回到金属阳离子的价态电荷越多,使得Nb-O 和Bi-O间的杂化越强,从而达到降低其总能的效果。

图5 ABi2Nb2O9体系中的Bi 6s,6p和Nb 4d态的分态密度
2.3 化学键
金属阳离子与O2-的2p态杂化使它们之间存在明显的共价键,而这种共价作用是导致ABi2Nb2O9体系铁电晶格失稳的主要原因。为了研究ABi2Nb2O9体系相变过程中化学成键的变化及离子在成键过程中电荷迁移的变化,对电荷密度及结合Bader的原子分子理论(AIM)进行了分析。顺电相(图6(a))和铁电相(图6(b))的电荷密度在[100]方向上的分布如图6所示。NbO6八面体内的Nb原子与相邻的O原子(这里只显示O(1), O(2)和O(4))及Bi2O2层的Bi原子和O(3)之间呈现出明显的电荷分享,表明它们之间存在强烈的共价作用。A位离子与相邻O2-之间尽管表现出离子键的特征,但是少量电荷之间的分享,说明它们之间也存在共价键的作用。相比顺电相来说,一方面,铁

图6 ABi2Nb2O9顺电相和铁电相在[100]方向上的电荷密度分布Fig.6 Charge density distribution of ABi2Nb2O9paraelectric and ferroelectric phases in [100] direction
电相的NbO6八面体之间显示出更强的电荷分享,Nb-O共价键加强,有利于铁电相变;另一方面,Bi2O2层与O(2)间相互吸引导致了NbO6八面体的畸变,这类似于以往SBT的报道中,Bi-O(2)之间的相互作用导致了晶格失稳,促进铁电畸变的发生。
电荷的成键实质上反映了离子间电荷的转移和重新分布的过程,电荷迁移计算的结果见表2,表中名义上的电荷表示在纯离子材料中对应原子的电荷迁移量。结果表明:ABi2Nb2O9体系原子电荷均少于在纯粹离子化合物中名义上的电荷(A为+2 eV,Bi为+3 eV,Nb为+5 eV,O为-2 eV),其中Bi(大约为1.88 eV),Nb(2.66 eV左右) 和O(在-1.13 eV到1.31 eV范围内)离子远小于名义的电荷,这也反映了Nb-O和Bi-O之间的强烈杂化使部分O的价态电荷返回到Nb5+离子和Bi3+离子,使它们之间存在很强的共价作用。根据COHEN的观点,长程的库仑力有利于铁电相,短程的排斥力有利于顺电相,因此,Nb-O和Bi-O之间具有强烈的共价作用,削弱了短程排斥力的作用,有利于ABi2Nb2O9铁电相的稳定。另一方面,A位离子的不同也导致了ABi2Nb2O9各离子间电荷迁移上的差异,集中体现在Nb5+,O(1),O(4)和O(5)电荷随A位离子的不同而原子内电荷有所不同。而Bi3+,O(2)和O(3)原子电荷的迁移保持一个相对较稳定的量,这说明A位离子主要作用于相邻O2-离子,Bi2O2层的O(3)及远离A位离子八面体上的O(2)受A位离子的影响较小。PBN中Pb2+电荷(1.40 eV)远小于CBN中的Ca2+电荷(1.62 eV),这与较大Pb-O间的杂化和相对最小Ca-O间的杂化一致。因此,电荷迁移的多少定性反映了离子间的成键强度。
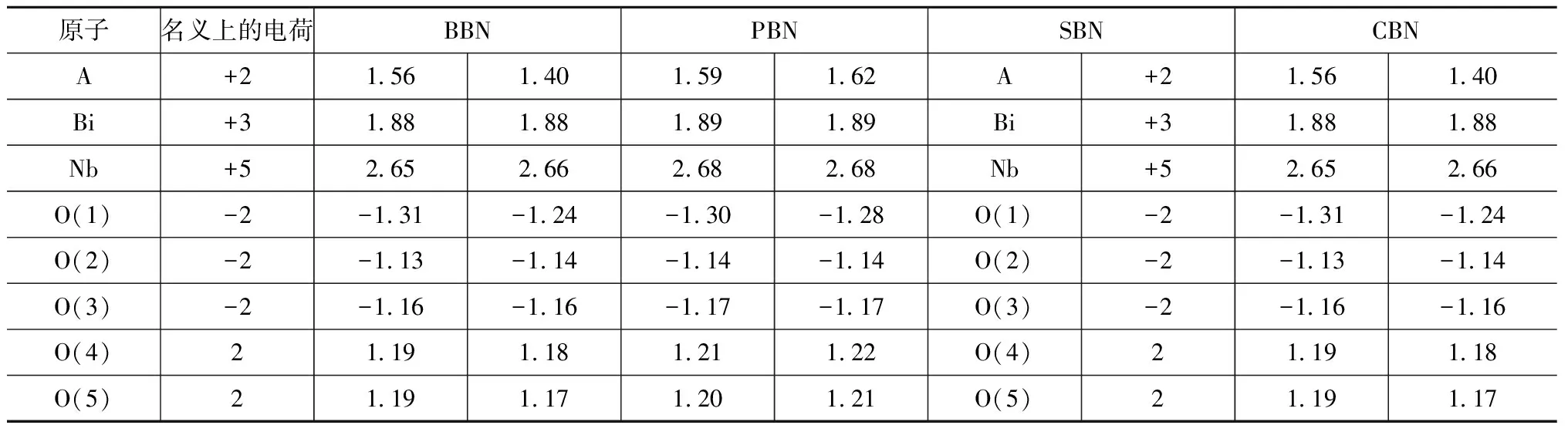
表2 ABi2Nb2O9 (A=Ba,Pb,Sr,Ca)原子basin内的电荷迁移Tab.2 Migration of charges in basin of atoms of ABi2Nb2O9 (A=Ba, Pb, Sr and Ca) eV
综上可知,铁电相变由原子间的共价键、长程库仑力及离子间短程排斥力等因素间微妙的平衡共同决定。
2.4 极化强度方案
在ABi2Nb2O9体系自发极化的计算中,采用能够精确计算相关离子位移极化的现代极化理论方法,即Berry-Phase理论方法。在计算过程中遵循Born-von Karman 周期性的边界条件,因此总的极化包含了一个任意的极化量∣eR/Ω∣,此处e为电子的电量,R为极化方向上的晶格矢量,Ω为晶胞的体积。
对于ABi2Nb2O9体系,沿a轴方向上的极化是从四方的顺电相向正交铁电相结构转变过程中离子沿a轴方向的位移。考虑到相变过程中存在一个非极化的正交结构,极化可被认为是非极化的正交结构向铁电相转变直接导致的,尽管部分ABi2Nb2O9体系的中间过度相在实验中未被报道,但从四方结构向非极化结构转变过程中并不会引入极化,因此这不影响把非极化的正交结构作为参考相来计算ABi2Nb2O9体系极化的准确性。
ABi2Nb2O9体系总的极化等于(Pmin+n∣eR/Ω∣)μC/cm2,a为[100]方向上的晶格矢量,其中最小极化量Pmin和晶格极化量∣eR/Ω∣的计算结果见表3。比较n=0时ABi2Nb2O9体系的极化Pmin,可发现A位离子对Pmin的影响较大。

表3 基态铁电相体系沿[100]方向上最小极化量Pmin和晶格极化量∣eR/Ω∣Tab.3 Minimum polarization value Pmin and lattice polarization value∣eR/Ω∣ of ground state ferroelectric phase ABi2Nb2O9 system in [100] direction μC/cm2
从BBN到CBN,Pmin随A位离子半径的减小而增大,而PBN显示出最大的Pmin(8.2 μC/cm2)主要是由于Pb2+活跃的孤立电子对的作用,BBN很小的Pmin(0.2 μC/cm2)反映了A位的Ba2+抑制了NbO6的畸变,这与表1显示的畸变参数是一致的。结果表明极化与A位离子的关系为:1) A位离子半径越小,提供给系统畸变的活跃空间越大,越有利于系统大的自发极化;2) A位离子含有活跃的孤立电子对(如Pb2+, Bi2+),A-O之间强烈杂化导致体系大的畸变,有利于更大的自发极化。在改善SBN薄膜的极化时,选用尺寸小或具有孤立电子对的Pb2+, Bi2+等作为A位离子对SBN进行部分取代或者掺杂,可提高其极化强度。4种体系计算结果如图7(a)所示,ABi2Nb2O9体系的自发极化随晶格极化量|eR/Ω|的倍数n的变化关系反映了[100]方向上的极化与晶格矢量a的关系,由于ABi2Nb2O9体系的晶格常数比较接近,所以对应的自发极化的增量基本相同。对于不同的n值对应的自发极化P,反映了ABi2Nb2O9薄膜生长过程中有可能出现的极化,这说明ABi2Nb2O9薄膜的极化并不唯一的稳定在某一确定的极化值。选择不同的晶格取向、生长气氛和退火条件,都将可能改变ABi2Nb2O9薄膜的自发极化,即对应着不同的n值。SBN有关极化的实验结果和本次理论计算的结果对比如图7(b)所示,结果表明实验中出现的极化基本上与理论的计算接近。

图7 ABi2Nb2O9体系的极化P随晶格极化量倍数n的变化Fig.7 Changes in polarization P of ABi2Nb2O9 system along with coefficient n of lattice polarization value
3 结论
本文综合分析了ABi2Nb2O9(A=Ba, Pb, Sr, Ca)体系的结构、电子属性、化学键和极化属性。电子结构和化学成键的分析结果表明了ABi2Nb2O9体系有着类似的铁电起源,Nb-O和Bi-O间的杂化在铁电相变的过程对于系统的畸变和铁电相的稳定起着至关重要的作用。A位离子的半径强烈影响着它们的带隙和结构的畸变。随着A位离子从Ba2+变化到Ca2+,带隙从2.10 eV 增加到2.67 eV,NbO6八面体的畸变参数也从3.12×10-3增加到3.42×10-3,但是PBN中活跃的Pb2+孤立电子对加强了PBN的结构畸变,使其相比其他的ABi2Nb2O9体系有更大的畸变参数,具有更大的自发极化,所以A位离子为Ca2+离子的材料更适合作为铁电移相器材料。
猜你喜欢
沈阳建筑大学学报(自然科学版)(2022年4期)2022-11-15
实验室研究与探索(2022年7期)2022-10-26
无机化学学报(2022年9期)2022-09-16
金属热处理(2022年3期)2022-04-09
井冈山大学学报(自然科学版)(2022年2期)2022-03-31
照相机(2020年9期)2020-10-28
中学生数理化(高中版.高考理化)(2020年10期)2020-10-27
新高考·高一物理(2016年7期)2017-01-23
读写算·教研版(2016年8期)2016-05-07
新高考·高一物理(2015年6期)2015-09-28
