
Cockayne综合征患儿临床特征及基因突变研究一例报告
2019-01-08 03:05:14潘璐刘艳秋
江西医药 2018年11期
潘璐,刘艳秋
(江西省妇幼保健院产前诊断中心,南昌 330006)
科凯恩综合征(Cockayne syndrome,CS)是一种罕见的常染色体隐性遗传疾病,于1936年由英国儿科医生Cockayne[1]首次对该疾病进行报道。在美国及欧洲国家该病的发病率大约是2.7/1000000[2],国内偶见散发个例报道。Coakayne综合征是一种以身材矮小、早衰外观为特征,该病特点包括体重不增、不能正常生长、头围异常小和神经系统发育受损。患儿对光极度敏感,少量阳光照射就能引起晒伤,其他症状包括听力丧失、眼部异常、蛀虫严重、骨质异常并有脑部特征性变化[3,4]。目前已知的主要致病基因有 ERCC6、ERCC8,ERCC8位于人类第5号染色体 (5q12.1),ERCC6位于人类第10 号染色体(10q11.13)[5],另外少见的 XPB、XPD、XPG基因亦可导致Cockayne综合征[6]。本研究中,我们对1例Cockayne综合征患儿应用芯片捕获高通量测序及其父母采用Sanger测序法对ERCC6、ERCC8进行突变测序,明确其突变类型及突变形式,并根据遗传学规律指导该夫妇避免再次生育Cockayne综合征患儿。
1 对象与方法
1.1 对象 江西省妇幼保健院就诊患儿,男,5岁,足月分娩,分娩时无窒息、缺氧等,1岁内生长发育基本正常,2岁开始生长发育增长缓慢,并出现运动、智力发育迟缓,少量阳光照射即可出现晒伤、脱皮、色素沉着,3岁时能独立行走,步态不稳,并出现睾丸萎缩,眼球凹陷,间断出现腹泻,5岁时睾丸消失,现只会说简单的词句。该患儿为第二胎第二产,足月顺产,出生体重2.6kg,身长50cm,Apar评分9分,生后母乳喂养。专科检查:身高85cm,体重10kg,颜面部皮肤增厚、粗糙、脱屑,有色素沉着,眼球凹陷,头发稀疏,神志清楚,表情淡漠,心肺未闻及明显异常,肝脾稍偏大,睾丸消失。实验室检查:染色体 G 显带:46,XY;血常规、血凝、生化、自身免疫、血气分析、病原体及病毒检测等未见明显异常。头颅CT提示:颅内广泛性钙化。患儿父母非近亲婚配,无遗传家族病史。
1.2 方法
1.2.1 DNA提取 用含枸橼酸钠抗凝剂的真空采血管,分别采集患儿及其父母外周静脉血5ml,应用试剂盒 (采用美国OMIGA公司E.Z.N.A Blood DNA试剂盒)提取DNA,提取步骤参照试剂盒说明;取适量DNA用紫外分光光度进行定量和纯度检测,其余保存于-20℃备用。
1.2.2 基因芯片捕获高通量测序 应用基因芯片(华大基因有限公司)对靶基因进行捕获,对捕获的富集靶基因采用高通量测序(在Hiseq2000测序平台进行测序--美国 Illumina公司)对 ERCC6、ERCC8基因进行突变检测,采用Illumina配套软件进行信息处理和数据分析。
1.2.3 Sanger验证 利用Primer软件对目标位点进行特异引物设计(正向引物序列5,-GAGACTCCCA CTTCTCATACTCATAAAGGGGTGT-3’反向引物序列 5’ -GTCTTTGTCCTCTTTTTCGGCTTTTTC-3’)(引物由华大基因股份有限公司合成),对该患儿基因组DNA进行目标位点及上下游区域进行扩增;扩增产物采用天根通用DNA纯化试剂盒进行纯化;纯化产物用Beckman 8000型DNA测序仪进行测序;测序结果与基因库的ERCC6基因参考序列 (NC-000010.10)进行比对,进一步确认核苷酸变异信息。
1.2.4 突变分析 测序结果用Chromas软件分析,并与NCBI中的正常序列进行BLAST比对,分析采用在线软件分析蛋白的结构,检测保守区、功能区及多序列比对。在线分析软件为:0nline Mendelian Inheritance in man (OMIM)、1000 Genomes、PolyPhen 一 2、SIFT、SWISS.Model repository、PROSITE。
2 结果
测序结果提示患儿存在ERCC6基因复合杂合突变(表 1),Sanger测序(表 2):对父母进行同片段序列分析结果显示,该患儿分别遗传了父亲及母亲的杂合突变,该患儿ERCC6复合杂合突变中核苷酸 变 化 c.116-1125delTGAGTATTTC、c.780-781insCC; 氨基酸变化:p.Ser372SerfsX30|p.S372SfsX30、p.ProfsX70|p.P260PfsX70; 基因亚区:EX5;CDS4,属框移突变,该框移突变导致氨基酸编码蛋白发生提前终止,产生截短蛋白,会对蛋白质的结果和功能产生较大影响;该突变在文献中未见报道;与CS发病有相关性。
3 讨论
CS是一组进行性加重的中枢神经系统遗传变性疾病,其发病机制是ERCC6、ERCC8基因发生突变,细胞内DNA受到损伤后,由ERCC6基因编码的CSB不能将损伤的DNA进行修复,并且以损伤的DNA作为模板进行mRNA转录并进行蛋白质翻译;ERCC8基因编码的CSA蛋白亦不能修复损伤的DNA,并且导致损伤的DNA累积,后续的转录及翻译过程终止,因此CS的临床表现可能是修复缺陷与转录缺陷共同作用的结果[7,8]。

表1 患儿ERCC6基因突变位点信息

表2 患儿父母Sanger验证位点信息

图1 (图1a为该患儿c.780-781insCC突变位置、图1b为该患儿c.116-1125delTGAGTATTTC突变位置)
该患儿出现的临床症状与报道的CS综合征患者的临床表现相同,但该患者还出现了腹泻,患儿还有一哥哥,其哥哥与该患儿的症状一致,亦出现腹泻,并且有肝衰竭(11岁时已死亡)。Cockayne综合征有三种临床分型[9]:1 型(OMIM:133540)为经典型,累及脑、眼、皮肤、骨骼等多个系统,出生时多正常,婴儿期或儿童早期发病,逐渐进展;2型(OMIM:216400)为先天型,表现更严重,出生早期就可能死亡;3 型(OMIM:216411)为温和型,特点是晚发病,病程进展缓慢,可以存活至成年,通过该患儿的临床表现提示为Cockayne综合征1型。目前国内外还未见Cockayne综合征1型生育后代报道[10]。
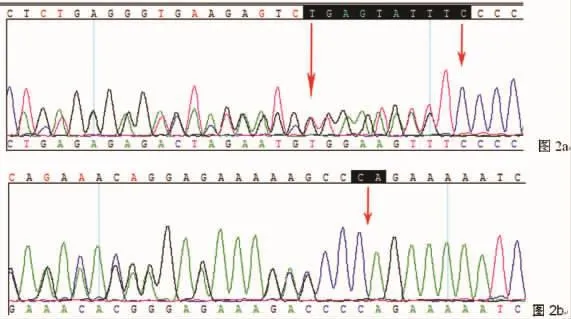
图2 (图2a该患儿母亲c.116-1125delTGAGTATTTC突变位置;图2b为该患儿父亲c.780-781insCC突变位置)
Cockayne综合征头部影像学检查最具特征性[11],表现为双侧侧脑室扩大、脑萎缩和钙化、脑白质髓鞘形成不良等,该患儿头颅CT提示颅内广泛性钙化,符合其影像学改变。Cockayne综合征没有血生化以及代谢产物的特征性改变,因此主要通过临床表现及影像学的特征性进行临床诊断,再通过基因检测进行分子遗传学确诊[12]。
本研究中,我们对该患儿进行了ERCC6、ERCC8基因进行检测,患儿检测出了ERCC6基因的 c.116-1125delTGAGTATTTC(p.Ser372SerfsX30)及 c.780-781insCC(p.ProfsX70)突变,属于复合杂合突变。根据Sanger测序,c.116-1125delTGAGTA TTTC杂合突变来源于父亲,c.780-781insCC杂合突变来源于母亲,目前该突变国际上未见报道,属于新发突变。患儿及其哥哥均为Cockayne综合征患者,其父母表型正常,但为致病基因携带者。Cockayne综合征是一种严重致残致死性遗传疾病,目前没有有效的治疗办法,只能采取物理及支持治疗提高患者的生活质量,因此患该病儿给社会及家庭带来了沉重的经济及精神负担,及时的诊断及产前诊断是非常有必要的。该夫妇再次妊娠后患病风险1/4,致病基因携带者1/2,鉴于该夫妇已生育2个该病患儿,因此在妊娠11-14周采用胎儿绒毛膜绒毛取样(CVS)方法或18-24周采用羊膜腔穿刺术进行产前诊断,或者针对该检出的致病基因位点直接行胚胎遗传前诊断(PGD),以阻止此病在该家庭中继续蔓延。
猜你喜欢
上海金属(2021年6期)2021-12-02 10:47:20
昆明医科大学学报(2021年3期)2021-07-22 07:40:04
种子(2021年3期)2021-04-12 01:42:22
生物学通报(2019年3期)2019-02-17 18:03:58
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
法医学杂志(2015年4期)2016-01-06 12:36:36
法医学杂志(2015年4期)2016-01-06 12:36:36
华东理工大学学报(自然科学版)(2014年1期)2014-02-27 13:48:29
当代畜禽养殖业(2014年6期)2014-02-27 07:59:03
赤峰学院学报·自然科学版(2013年4期)2013-07-31 22:01:40
