
试论毛细管气相色谱法测定食品中的甜蜜素
2019-01-03 07:19◎周琦
现代食品 2018年20期
◎ 周 琦
(费县检验检测中心,山东 临沂 277700)
甜蜜素的化学名称为环己基氨基磺酸钠,属于水溶性甜味剂,其甜度是蔗糖的50倍左右,且价格低廉,但摄入过量会导致人体肝肾系统受损,不利于人体健康。现阶段,在我国的食品加工中允许适度应用甜蜜素。但一些厂家为了改善食品的口感,会超范围、超剂量的应用甜蜜素。通过毛细管柱气相色谱检测食品中甜蜜素的含量,效果精准,可提升甜蜜素检测效果。
1 材料与方法
1.1 试剂
正庚烷[CH3(CH2)5CH3]、氯化钠(NaCl)、石油醚(沸程为30~60 ℃)、氢氧化钠(NaOH)、硫酸(H2SO4)、亚铁氰化钾{K4[Fe(CN)6]·3H2O}、硫酸锌(ZnSO4·7H2O)、亚硝酸钠(NaNO2)。
1.2 仪器与设备
气相色谱仪:配有氢火焰离子化检测器(FID);涡旋混合器、离心机(转速≥4 000 r·min-1)、超声波振荡器、样品粉碎机、10 μL微量注射器、恒温水浴锅、天平。
1.3 方法
1.3.1 样品前处理
在进行毛细管气相色谱法测定食品中的甜蜜素过程中,要进行样品前处理,其具体方式如下:①液体试样处理。将其摇均之后直接称取10 g样品,将其后置于50 mL具塞比色管之中。②固体试样。糕点以及面包类的制成粉状;而蜜饯、果脯等食品则通过粉碎机打磨形成粉状或均匀浆状液体,然后称取2.0 g放置在研钵之中,加入少许海砂,研磨成干粉状,利用漏斗导入到100 mL容量瓶,同时用水冲洗研钵,并将冲洗液转移到容量瓶中,然后加水摇动,1 h后过滤获得试样,精准吸取10 mL置于50 mL具塞比色管。
1.3.2 样品衍生以及提取
将样品混合均匀之后,将50 mL具塞比色管放置在冰水浴中,加入2.5 mL亚硝酸钠溶液,溶液浓度为50 g·L-1;再加入 2.5 mL 硫酸溶液,溶液浓度为 100 g·L-1,摇匀;然后在冰水浴中放置30 min后再次摇动,加入5.0 mL正庚烷、2.5 g氯化钠,摇匀后通过旋涡混合器振动1 min,或振摇80次,静置、分层后将正烷层析出,在10 mL的带塞离心管中对其进行离心,将上层正庚烷通过气相色谱仪进行分析处理。
1.3.3 样品的测定分析
可将衍生的以及提取好的正庚烷提取液进样通过1 μL在气相色谱仪进行分析。
1.3.4 配制标准系列
精准称取1.0 000 g的甜蜜素标准物质,加水溶解到定容为100 mL,将其配制形成浓度为10 mg·kg-1的标准溶液。取8支50 mL具塞比色管,分别加入10 mL水,然后再分别加入 0、0.002 5、0.012 5、0.05、0.10、0.25、0.50 mL和1.00 mL标准溶液,分别对应0、5、25、100、5 000、500、1 000 μg和2 000 μg的标准物质。
2 结果与分析
2.1 样品预处理优化分析
将普通液体试样摇匀后,称取25.0 g试样样品,根据需求确定是否要进行过滤处理,通过水定容为50 mL备用。
称取25.0 g试样放置于烧杯中,通过氢氧化钠溶液进行处理,调整pH至7~8,60 ℃水浴加热30 min,清除酒精后放冷,用水定容到50 mL备用。
2.2 固体、半固体试样的处理
处理低脂、低蛋白样品,先称取打碎后混匀的样品3.00~5.00 g置于50 mL离心管中,加入30 mL水,振摇并超声提取20 min,混匀后再离心(3 000 r·min-1)10 min后过滤,分次洗涤残渣,收集滤液并定容至50 mL备用。
将高蛋白样品放置于在250 mL烧杯,融化后搅拌均匀,称取3.00~5.00 g置于50 mL离心管中,加入30 mL水,振摇并超声提取20 min,再加入2 mL亚铁氰化钾溶液,混匀后加入2 mL硫酸锌溶液,离心处理,通过3000 r/min的速度离心10 min后过滤,分次洗涤残渣,收集滤液并定容至50 mL备用。
2.3 条件优化手段分析
2.3.1 色谱分析
国家标准GB 5009.97-2016中应用气相色谱分离甜蜜素。色谱柱为弱极性石英毛细管柱,在管住内部涂5%苯基甲基聚硅氧烷,30 m×0.53 mm×1.0 μm或等效柱。分别吸取1 μL,通过衍生化处理标准系列各个浓度的清液,再注入气相色谱仪中,测得不同浓度被测物产生的响应值峰面积,基于浓度作为横坐标,环己醇硝酸以及环己醇两峰面积相加获得的和作为横坐标,绘制标准曲线。
在相同的条件下进样1 μL,通过衍生化出流后的试样,通过待测液体上清液,获得峰面积结果,综合标准获得组分浓度,如果清液响应值高于线性范围,再通过正庚烷稀释在对其进行分析。
2.3.2 柱温升温程序
初始温度为55 ℃,保持时间为3 min,以10 ℃/min的速度升温到90 ℃,且保持0.5 min,然后在以20 ℃/ min的速度将其升温到200 ℃,并保持3 min。
2.3.3 进样口
进样口的温度为230 ℃,其进样量为1 μL,不分流/分流进样,分流比为1∶5。而分流比和方式均可基于具体的色谱仪器条件进行合理调整和优化。
2.3.4 优化标准曲线制备
应用国标要求,进样1~5 μL绘制标准曲线,在标准浓度出现变化的时候,如果存在操作失误则就会造成整体曲线出现变化,这种变化不易发现。而因为正庚烷具有一定的挥发性,样品在进样口的时候很容易气化,而进样量的差异会导致样品在进样口气化体积出现一定差异,因此无法获得精准的浓度数值。如果标准管在实验中出现的误差则就会直接影响实验结果的精准性。在实验过程中,可以通过配制5个不同浓度的甜蜜素标准系列进行衍生,通过回归计算的方式抵消误差,保证试样进样量以及标准进样量的相同性,使操作更加科学,结果更加精确。
2.4 线性范围以及检出限
分别吸取1 μL衍生化处理标准系列中不同浓度的溶液,将其注入到气相色谱仪器中,可以测量5个浓度被测物的响应值峰面积,将浓度设为横坐标,将对应的峰面积设为纵坐标,绘制标准的曲线,相关系数r=0.999 92。采用毛细管柱法测定的标准浓度的线性范围是5~2 000 μg/mL,根据3倍基线确定检出限浓度数值为1.5 μg/mL;液体取样10 g,方法检出限数值为0.8 mg/mL,方法定量限为2.5 mg·kg-1;固体取样10 g,处理液定容体积为100 mL,将10 mL的衍生化计,此种方式检出限设为 8 mg·kg-1,方法定量限为 25 mg·kg-1。
2.5 方法精准度以及精密度、回收率的测定分析
通过对蜜饯与饮料的样品中加入标准曲线方位中不同浓度的标准溶液,将每个浓度平均分为6份,连续6 d,根据样品处理的方式进行实验处理,对日内以及日间精密度、绝对回收率进行分析。其中相对标准偏差要控制在1.19%~4.09%,绝对回收率为90.0%~104.0%。
2.6 样品分析结果
分别将红葡萄酒、蜜饯以及饮料中随机选择10份样品测定甜蜜素含量,将衍生以及提取完成的正庚烷提取液进行分析,结果见表1。
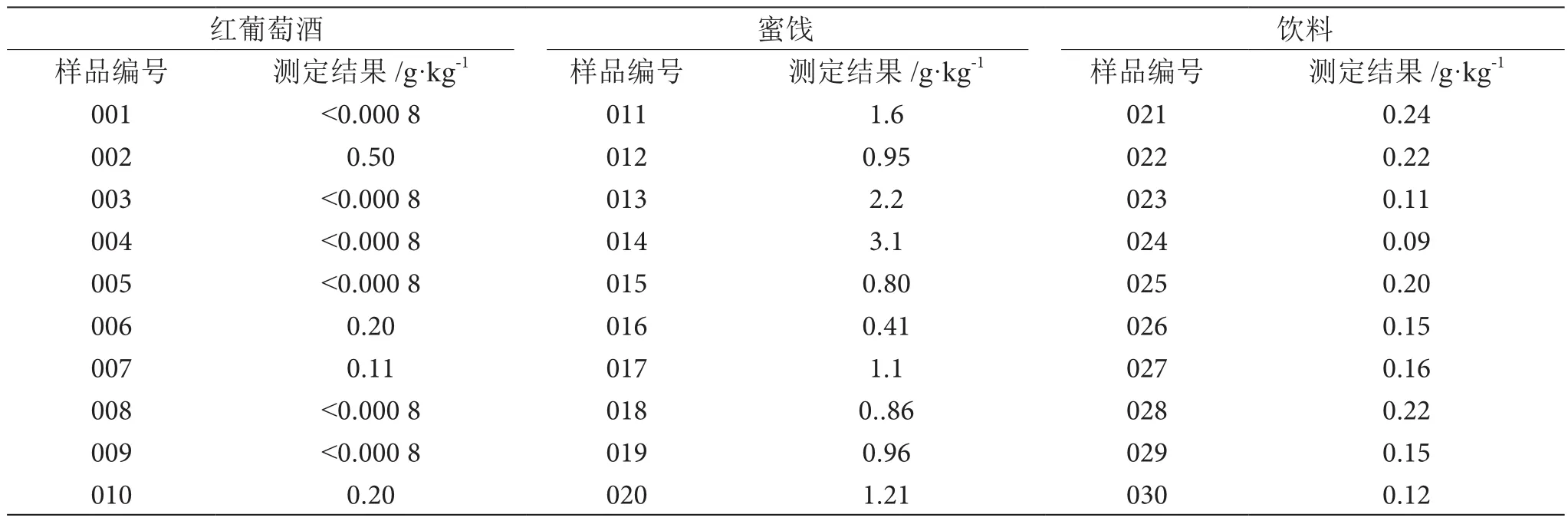
表1 10份样品的甜蜜素含量测定表
3 结论
通过萃取、衍生,提取样品后利用毛细管柱气相色谱仪对食品中的甜蜜素进行测定,具有高分辨率、高灵敏性、低检出限以及测定速度快等显著优势,现阶段应用较为广泛,是一种较为精准的检测方式。随着科学技术手段的不断提升,甜蜜素的测定方式势必会更加灵敏、便捷,应用范围也会更为广泛。
猜你喜欢
化工设计通讯(2022年10期)2022-12-31
波谱学杂志(2022年2期)2022-06-14
工程技术与管理(2022年7期)2022-03-04
哈尔滨工业大学学报(2020年1期)2020-12-21
化工设计通讯(2020年11期)2020-01-12
中国市场(2016年36期)2016-10-19
浙江大学学报(工学版)(2016年11期)2016-06-05
广西农学报(2015年2期)2015-10-14
郑州大学学报(工学版)(2014年6期)2014-03-01
