
长链非编码RNA BRE-AS1慢病毒表达载体的构建与鉴定
2018-11-29 06:10周辉余淦徐华叶章群
现代泌尿生殖肿瘤杂志 2018年5期
周辉 余淦 徐华 叶章群
长链非编码RNA(long noncoding RNA, lncRNA)是生物体产生的一类不具有蛋白编码功能的RNA转录本,其通常序列大于200 bp。此类RNA分子在以往被认为是“转录垃圾”,但目前越来越多的研究显示,lncRNA在细胞增殖、凋亡、迁移、侵袭等生物学进程中都发挥着举足轻重的作用,也在包括肿瘤在内的多种人类疾病的发生、发展中发挥重要调控作用[1-3]。BRE-AS1又名NR_028308,位于人类染色体2p23.2,是一种新发现的lncRNA分子,序列长度为1 667 bp。BRE-AS1为BRE基因的反义RNA,我们前期的lncRNA表达谱研究显示BRE-AS1在肾细胞癌组织中显著低表达,但其在肾细胞癌中的生物学功能及具体的分子机制仍不明确[4]。为了对其生物学功能及机制进行进一步研究,我们需要构建其表达载体。慢病毒载体以人免疫缺陷Ⅰ型病毒(HIV-1)的结构基因为基础,可将目的基因整合到宿主细胞的基因组上,可高效率、持续、稳定地表达外源性基因;此外,慢病毒对分裂期及非分裂期细胞均具有较强感染力,且具有较低免疫原性,通常不会对细胞活性造成较大影响[5-6]。由于这些优势,慢病毒表达系统在生物学研究中得到了越来越广泛的应用。本研究通过构建BRE-AS1基因的慢病毒表达载体,为进一步研究BRE-AS1在肾细胞癌中的生物学功能及分子学机制打下基础。
材料与方法
一、材料
慢病毒表达质粒系统包括载体穿梭质粒pCDH-CMV-MCS-EF1-puro-copGFP、慢病毒结构质粒pPACKH1质粒体系(pPACKH1-GAG、pPACKH1-REV、pPACKH1-VSVG),购自美国SBI公司。转染试剂为瑞士罗氏公司生产的X-tremeGENE HP DNA Transfection Reagent,慢病毒病毒沉淀剂PEG6000购自上海吉凯基因化学技术有限公司。DH5α感受态大肠杆菌购自武汉翰林博公司。质粒小提试剂盒购自美国OMEGA公司,琼脂糖凝胶回收试剂盒购自北京天根生物技术有限公司;胎牛血清、DMEM高糖培养液、RPMI-1640培养液、胰酶、PBS缓冲液购自美国Hyclone公司;TRIzol试剂、KOD-plus高保真聚合酶、逆转录试剂盒、实时定量PCR试剂盒、限制性内切酶分别购自美国Invitrogen、日本TOYOBO、日本TAKARA、瑞士Roche、美国Thermo公司。引物合成及DNA鉴定由武汉擎科生物公司完成。EdU细胞增殖检测试剂盒购自广州锐博生物科技有限公司。
二、实验方法
1.重叠延伸PCR法获取BRE-AS1目的序列:根据NCBI检索,确定BRE-AS1的序列,设计引物:扩增BRE-AS1序列全长的引物BA-F:5′-CGGAATTCGTATGATTCCCCTCATCAGGCA-3′(含EcoR Ⅰ酶切位点及保护碱基),BA-R:5′-ATTTGCGGCCGCTTTGGAATATCTGAATTTATTACT-3′(含Not Ⅰ酶切位点及保护碱基)。 提取293TN细胞的总RNA逆转录获得cDNA作为模板,利用KOD-plus高保真聚合酶进行PCR。由于前后引物解链温度(Tm)差别较大,未能得到正确的PCR产物,我们设计了新的PCR引物,进行重叠延伸PCR反应。用于重叠延伸PCR的引物序列:BA-F1:5′-CTGTCACCTTTCACCATGTTGATTGTCGGCA-3′,BA-R1:5′-TGCCGACAATCAACATGGTGAAAGGTGACAG-3′。BA-F及BA-R1用于PCR得到产物为1 400 bp,BA-F1及BA-R用于PCR得到产物为311 bp;将PCR产物琼脂糖凝胶回收后混合作为模板,用BA-F及BA-R进行PCR得到产物长度为1 680 bp。此外,设计并合成BRE-AS1实时定量PCR前后引物,引物序列分别为F0 5′-AGGCAGTGGCAAGAGGAGGTG-3′及R0 5′-TCGGAGGTAAAATGACGTGGG-3′,PCR产物长度为431 bp。
2.重组BRE-AS1慢病毒载体质粒的构建与鉴定:将目的片段及pCDH-CMV-MCS-EF1-puro-copGFP载体分别由EcoR Ⅰ及Not Ⅰ内切酶进行酶切,并用琼脂糖凝胶电泳进行分离纯化,回收酶切的载体和目的基因片段。将酶切回收的载体DNA、目的基因片段、Ligation High连接酶混匀并孵育2 h,并转化到DH5α感受态大肠杆菌中,并接种到含氨苄青霉素的软琼脂平板。挑选菌落利用实时定量PCR引物进行PCR,得到阳性菌落后进行DNA测序。测序正确的菌落再用于质粒提取,用于后续实验。
3.慢病毒包装:根据慢病毒包装说明书,于6孔板中培养293TN细胞,细胞汇合度达到60%~80%时开始进行质粒转染。首先制备质粒转染混合物(6孔板每孔):于1.5 ml离心管中加入200 μl的opti-MEM培养液,依次加入1 600 ng的目的质粒及3种慢病毒结构质粒(各800 ng)后,加入10 μl转染试剂,混匀后孵育20 min,均匀加入293TN细胞的培养液中,摇晃混匀。置于37 ℃、5% CO2条件的细胞培养箱中培养8 h后换液,并继续培养96 h。收集293TN细胞上清液,以0.22 μm滤器过滤上清液。在病毒上清液中加入适量PEG6000病毒沉淀剂,于4 ℃沉淀过夜后,在离心机上以1 500 g速度离心30 min后可得到白色沉淀,弃上清保留白色沉淀,然后冰PBS液重悬病毒沉淀,并于4 ℃溶解过夜。
4.慢病毒滴度测定:采用稀释滴定法,将慢病毒溶液按10倍梯度稀释为原浓度的1/10、1/102、1/103、1/104、1/105、1/106共6个浓度梯度,分别感染接种293TN细胞。96孔板每个孔中加入90 μl培养液及10 μl稀释病毒液,各稀释梯度分别行3个复孔。72 h后利用倒置荧光显微镜计数各浓度梯度下有绿色荧光的细胞数,并取平均值。慢病毒滴度=细胞数平均值×稀释度×100(TU/ml)。
5.感染目的细胞及稳定过表达细胞筛选鉴定:将收集的BRE-AS1重组慢病毒和对照空病毒分别感染肾细胞癌细胞系OS-RC-2,72 h后用荧光显微镜观察转染效率。用嘌呤霉素(终浓度2 μg/ml)筛选带抗性细胞2周,得到稳定过表达目的基因的细胞。提取总RNA、逆转录、行荧光实时定量PCR,确定BRE-AS1的表达水平。
6.EdU插入实验:采用EdU细胞增殖检测试剂盒进行实验。首先将处理好的细胞种植到24孔板中,待细胞密度约70%~80%时,每孔加入稀释好的EdU培养液孵育2 h,清洗细胞后以4%多聚甲醛固定细胞,并用2 mg/ml甘氨酸终止反应。其后加入Apollo染色反应液,在室温下避光摇床孵育30 min,清洗后再加入Hoechst33342反应液进行DNA染色,清洗后在荧光显微镜下检测细胞的增殖能力。EdU为胸腺嘧啶核苷类似物,能掺入正在复制的DNA分子,与Apollo荧光染料反应能使增殖细胞的核产生红色荧光;而Hoechst33342能使所有细胞的细胞核产生蓝色荧光,因此带红色荧光与带蓝色荧光的细胞核的比例即为增殖细胞所占比例。
三、统计学方法
定量资料以均数±标准差表示,组间比较采用t检验进行分析;定性资料采用卡方检验进行分析。数据分析采用SPSS 19.0软件,P<0.05为差异有统计学意义。
结 果
一、BRE-AS1目的片段的重叠延伸PCR
扩增BRE-AS1序列全长的引物BA-F及BA-R用于PCR,未能得到预测长度的PCR产物,因此我们进行了重叠延伸PCR来得到BRE-AS1目的片段。与重叠延伸PCR的模式图(图1A)相一致,BA-F及BA-R1用于PCR得到产物大小约为1 400 bp,而BA-F1及BA-R用于PCR得到产物大小为311 bp(图1B);而将两种PCR产物分别琼脂糖凝胶纯化及胶回收后混合作为模板,用BA-F及BA-R进行PCR,得到产物长度为1 680 bp(图1C)。条带大小与预期各片段长度一致。
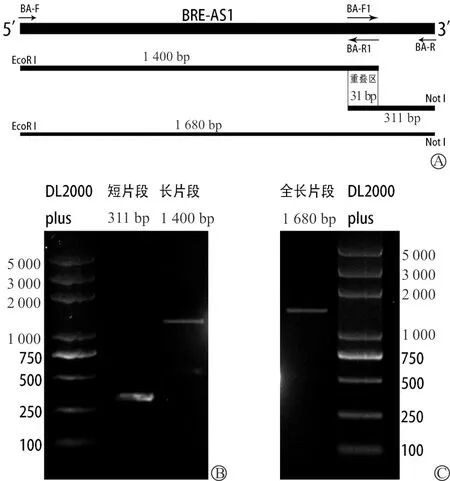
A:重叠延伸PCR的模式图;B:将PCR产物进行琼脂糖凝胶电泳,可见BRE-AS1长、短片段的大小与预期一致,短片段条带位于250~500 bp(大约311 bp),而长片段条带位于1 000~1 500 bp(大约1 400 bp);C:将长、短片段分别进行胶回收后混合作为模板进行重叠延伸PCR,获取BRE-AS1全长片段后进行琼脂糖凝胶电泳,可见一条明显的条带位于1 500~2 000 bp(大约1 680 bp)
图1 利用重叠延伸PCR进行BRE-AS1目的条带获取
二、pCDH-BRE-AS1载体的构建与鉴定
利用EcoR Ⅰ及Not Ⅰ内切酶,分别切割BRE-AS1目的片段及pCDH质粒,通过琼脂糖凝胶电泳及胶回收纯化目的片段后,依次进行连接、转化、挑菌落克隆、PCR鉴定、DNA测序。DNA测序正确的菌落,摇菌后进行质粒提取,用于后续实验。DNA测序结果与NCBI上BRE-AS1的RNA序列(NR_028308)进行比对,显示序列均一致(图2),表明重组载体构建正确。将该BRE-AS1表达载体质粒称为pCDH-BRE-AS1,空载体为pCDH-empty。
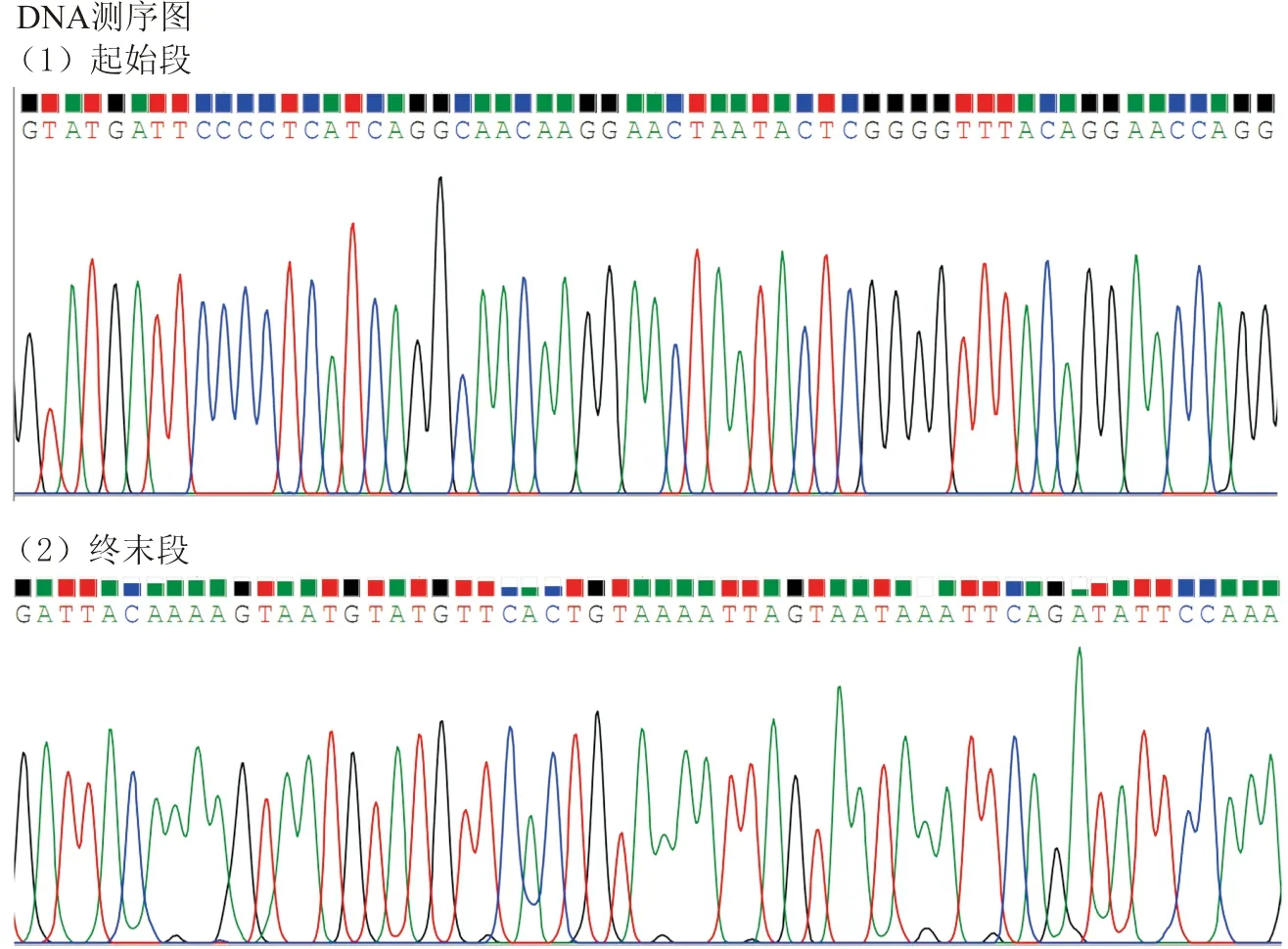
图2 pCDH-BRE-AS1载体的测序鉴定
三、重组慢病毒的包装及滴度测定
将pCDH-BRE-AS1转染293TN细胞获得病毒液,按梯度稀释后感染293TN细胞,根据公式计算出重组慢病毒的最终滴度。在本实验中BRE-AS1重组慢病毒的滴度为8.6×107TU/ml,BRE-AS1病毒液的合适稀释浓度为1/104倍。
四、感染目的细胞及稳定过表达细胞筛选鉴定
用过表达BRE-AS1的慢病毒液及空载体病毒液分别感染OS-RC-2细胞,72 h后即可见部分细胞存在绿色荧光。利用嘌呤霉素筛选抗药细胞2周后,得到稳定转染细胞;进行荧光显微镜观察,几乎所有细胞均呈现绿色荧光,转染效率接近100%(图3A)。此外,被感染的OS-RC-2细胞均未出现显著形态学变化或生长抑制,细胞状态保持良好。提取细胞总RNA、逆转录后进行荧光实时定量PCR,发现相对于转入空载体(pCDH-empty)的细胞,转染了目的序列(pCDH-BRE-AS1)的OS-RC-2细胞中,BRE-AS1的RNA表达水平增高约38.5倍(6.2个循环),差异有统计学意义(P<0.01)(图3B)。结果表明pCDH-BRE-AS1慢病毒表达载体能有效过表达BRE-AS1,该细胞可用于后续研究。
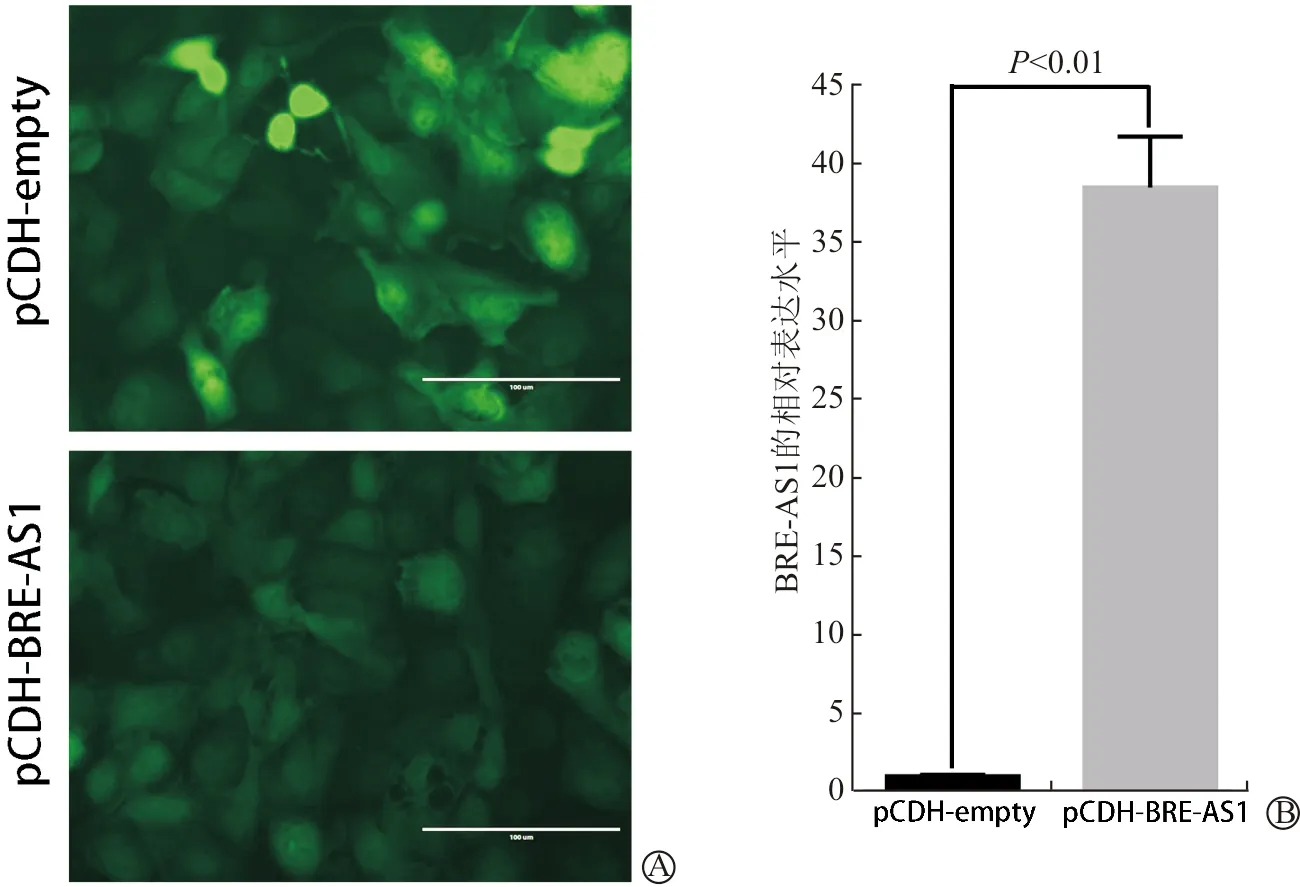
A:荧光显微镜观察(×200);B:荧光实时定量PCR检测BRE-AS1表达
图3 BRE-AS1慢病毒表达载体感染OS-RC-2细胞过表达BRE-AS1
五、EdU插入实验显示BRE-AS1抑制细胞增殖
将过表达BRE-AS1的OS-RC-2细胞及对照细胞种植于24孔板,利用EdU细胞增殖检测试剂盒进行EdU插入实验。图4A显示了两种细胞进行EdU插入实验后的细胞核染色情况。3个视野中,对照细胞中红色荧光细胞核与蓝色荧光细胞核的比例分别为43.75%、48.25%、40.35%,计算出对照细胞中约44.00%的细胞处于增殖状态;而稳定过表达BRE-AS1的OS-RC-2细胞中,3个视野中红色荧光细胞核与蓝色荧光细胞核的比例分别为28.93%、23.49%、25.36%,计算出该细胞中约26.00%的细胞处于增殖状态。相对于转入空载体的细胞,转染了目的序列(BRE-AS1)的OS-RC-2细胞中,增殖细胞比例显著下降,差异有统计学意义(χ2=70.3,P<0.01)(图4B)。结果显示BRE-AS1可抑制肾癌细胞的增殖,可能在肾癌的发生、发展中发挥抑癌作用。
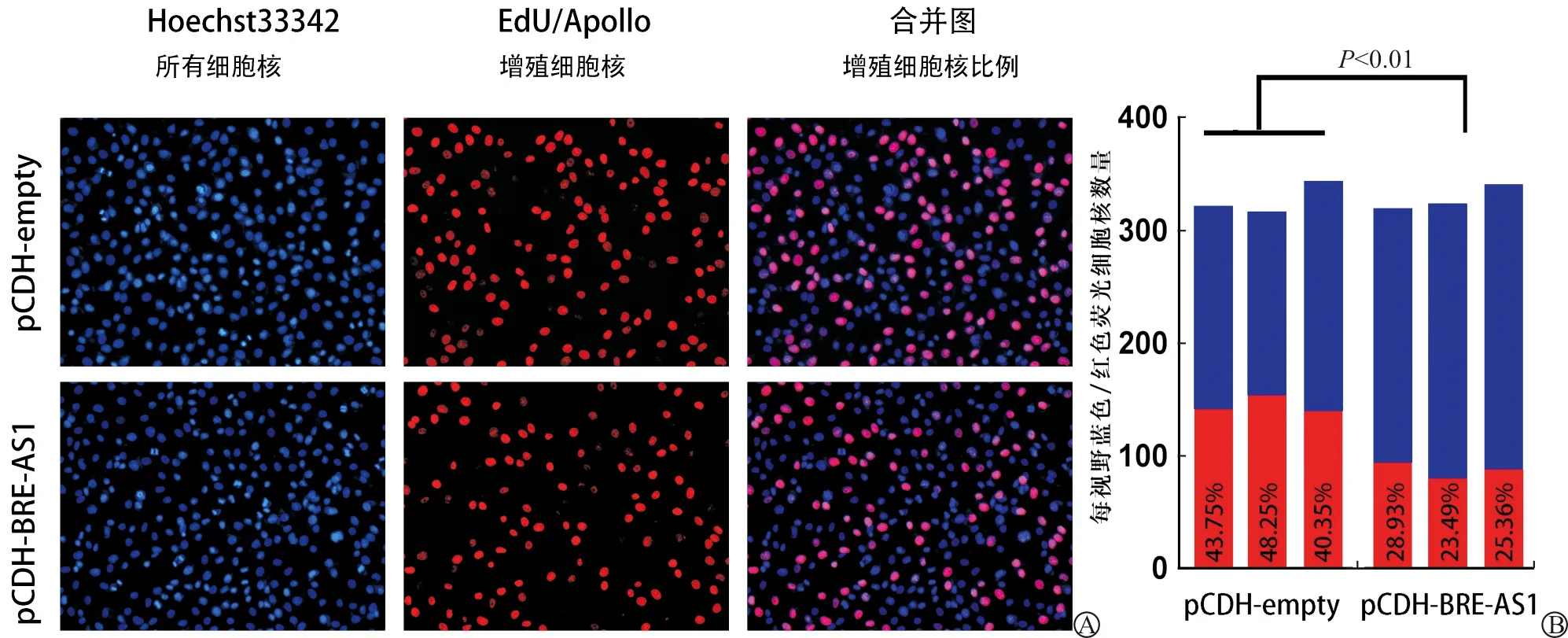
A:荧光显微镜显示,相对于对照细胞,在过表达BRE-AS1的OS-RC-2细胞中,红色荧光细胞核(增殖细胞)的比例较低;B:BRE-AS1过表达组增殖细胞所占比例显著降低
图4 EdU插入实验显示过表达BRE-AS1抑制肾癌细胞的增殖
讨 论
lncRNA能够调节多种生物学进程,参与多条信号通路并调控多种疾病的发生、发展,因此其在疾病中的作用越来越受到重视[2-3]。BRE-AS1为BRE基因的反义RNA,但是不具备蛋白编码功能,而是一种新的lncRNA。目前关于BRE-AS1的研究较少,张琳等[7]发现BRE-AS1在先天性小耳畸形患者的残耳软骨中表达水平显著上调,显示其与先天性小耳畸形的发生、发展有一定的关系。He等[8]则发现BRE-AS1在嫌色肾细胞癌中显著下调,且高表达BRE-AS1的患者具有更好的总生存期。此外,Pruunsild等[9]发现在培养的诱导性多能干细胞来源的小鼠神经元中,BRE-AS1的表达水平受突触活性调控,其可能参与神经冲动传导。在我们前期的肾细胞癌的lncRNA表达谱研究中,我们发现相对正常癌旁组织,BRE-AS1在肾细胞癌组织中显著低表达[4]。尽管如此,BRE-AS1在肾细胞癌中的生物学功能及其具体的分子机制,仍不为人所知,有待进一步研究。因此,构建其过表达载体就具有较为重要的意义。
慢病毒载体系统是一种以HIV-1的结构基因为基础的逆转录病毒载体系统,慢病毒载体系统具有一系列优势使其获得越来越多的关注和应用:慢病毒能较好感染分裂期及非分裂期宿主细胞;慢病毒载体可容纳较长基因片段;目的基因可在细胞内长期、稳定表达;免疫原性较弱,较少诱发宿主免疫反应,一般不引起细胞形态功能改变[5-6]。此外,慢病毒系统还能与最新的CRISPR/Cas9系统完美整合,高效在真核细胞中进行基因编辑[10]。为更好地研究lncRNA BRE-AS1在肾细胞癌中的生物学功能及其机制,我们尝试构建了BRE-AS1的慢病毒表达载体,以期高效安全的进行BRE-AS1过表达。本研究通过PCR得到BRE-AS1目的片段,将其重组到慢病毒载体中,成功构建基因表达载体质粒pCDH-BRE-AS1,并通过DNA测序鉴定表达载体质粒构建的正确性。随后我们通过慢病毒包装系统得到慢病毒并感染肾细胞癌OS-RC-2细胞系,利用嘌呤霉素筛选得到稳定表达BRE-AS1的肾细胞癌细胞。荧光显微镜下观察到转染效率接近100%,且感染后OS-RC-2细胞未出现明显的形态学变化,细胞生长状态及细胞活力保持良好。通过荧光实时定量PCR,我们证明转染了BRE-AS1慢病毒表达载体的OS-RC-2细胞中,BRE-AS1的RNA表达水平显著增加。这些研究结果显示,BRE-AS1慢病毒表达载体及稳定过表达BRE-AS1的肾癌细胞构建成功,这为后续进一步研究BRE-AS1在肾细胞癌及其他疾病的发生、发展中的生物学功能及潜在分子学机制,都奠定了良好基础。而后续的EdU插入实验也显示,过表达BRE-AS1能够降低增殖的肾癌细胞的比例,说明BRE-AS1能够抑制肾细胞癌的增殖,BRE-AS1可能在肾细胞癌发生、发展中发挥抑癌作用。这与其在肾细胞癌中显著低表达也较为一致。但后续还需要更多的体内及体外功能实验,来阐明BRE-AS1在肾细胞癌中确切的生物学功能。
此外,本研究的一个难点就是通过PCR得到BRE-AS1目的片段。BRE-AS1序列>1 500 bp,且其3′端序列中腺嘌呤(A)及胸腺嘧啶(T)的比例较高,利用头尾序列设计引物Tm值相差较大,进行PCR时无法得到目的序列,因此我们采用了重叠延伸PCR来获取目的片段。重叠延伸PCR也称为搭桥PCR,常用于获取大片段基因序列、基因定点突变、融合基因构建等,具有一系列优势[11-13]。本研究为获取BRE-AS1全长片段,将其分为前后两段,分别进行PCR后,将PCR产物混合作为DNA模板,再次进行PCR得到了正确的BRE-AS1目的片段。因此,为获取较长序列或前后引物Tm值悬殊的目的片段,重叠延伸PCR技术不失为一种值得尝试的好办法。
猜你喜欢
华人时刊(2022年9期)2022-09-06
江西农业学报(2021年4期)2021-04-20
水生生物学报(2021年1期)2021-02-04
中国生殖健康(2020年4期)2021-01-18
华人时刊(2020年15期)2020-12-14
三农资讯半月报(2020年11期)2020-06-21
中国现代中药(2019年5期)2019-07-03
科海故事博览·下旬刊(2019年6期)2019-04-16
中国生殖健康(2018年4期)2018-11-06
中国当代医药(2015年9期)2015-03-01
