
吉非替尼片的工艺研究和溶出曲线评价
2018-10-20 08:01王明森张菊红孙慧娟李大伟
食品与药品 2018年5期
王明森,张菊红,孙慧娟,李大伟,刘 杰*
(1. 山东省药学科学院,山东 济南 250101;2. 山东福瑞达医药集团有限公司 山东省黏膜与皮肤给药技术重点实验室,山东 济南 250101)
吉非替尼片由英国阿斯利康公司研制,商品名为IRESSA,于2007年在中国上市,治疗既往接受过化疗的局部晚期或转移性非小细胞肺癌(NSCLC),其中表皮生长因子受体(EGFR)基因敏感突变患者可用于一线治疗。它能与EGFR的三磷酸腺苷(ATP)激酶结合位点上的ATP 竞争,阻断其酪氨酸激酶活性,进而阻断 EGFR 的信号传导通路,阻碍肿瘤的生长、转移和血管生成,并可诱导肿瘤细胞的凋亡[1]。
吉非替尼易溶于冰醋酸、二甲基亚砜;溶于吡啶;难溶于四氢呋喃;微溶于甲醇、无水乙醇、乙酸乙酯、异丙醇和乙腈。按生物药剂分类系统(BCS),吉非替尼属于BCS 2类,即:低溶解性、高渗透性药物。本实验通过对吉非替尼片制备工艺关键点的研究,制备出合格的样品。依据仿制药一致性的评价指导原则,以体外溶出曲线拟合度为主要评价指标对吉非替尼自制片和参比制剂进行一致性评价[2]。
1 仪器与试药
1.1 仪器
SF-130B型万能粉碎机(上海天和制药机械公司);HLSG-10型湿法制粒机(北京国药龙立机械公司);ZPW-21B型旋转压片机(上海天和制药机械公司);LDCS型实验室高效包衣系统(USA Vector Corporation);Agilent 8453紫外-可见分光光度计(安捷伦科技公司);RCZ-8M溶出试验仪(天津市天大天发);电子分析天平XS-205(美国梅特勒-托利多公司)。
1.2 试药
参比制剂(英国阿斯利康公司,批号MC978);吉非替尼原料药(自制,批号170506);一水乳糖(Foremost);交联羧甲基纤维素钠(美国FMC);聚维酮K30(PVP K30,ISP);十二烷基硫酸钠(SDS,山河辅料);微晶纤维素(山河辅料);硬脂酸镁(山河辅料);胃溶型薄膜包衣粉(卡乐康);结晶乙酸钠(科密欧);磷酸二氢钾(国药集团);盐酸(科密欧);氢氧化钠(科密欧);冰醋酸(科密欧);纯化水(自制)。
2 方法与结果
2.1 吉非替尼单位制剂的处方组成[3]
吉非替尼250 mg,一水乳糖130 mg,微晶纤维素70 mg,羧甲基淀粉钠17 mg,聚维酮K30 20 mg,SDS 2.2 mg,硬脂酸镁4.5 mg,包衣增重2 %。
2.2 吉非替尼片的制备
(1)原料过200目,辅料过80目备用;(2)将原辅料混合均匀;(3)配制合适的PVP K30和SDS溶液,溶解完全,然后加入(2)中混合好的原辅料中制备软材,再用20目筛网制粒,60 ℃烘干,20目筛网整粒;(4)加入处方量的硬脂酸镁总混;(5)包衣。采用此工艺,制备3个批次(每批次2000片),批号为170201,170202,170203。
2.3 溶出曲线测定方法的建立
2.3.1 溶出介质选择和配制[4]吉非替尼溶解度与pH值相关,pKa值分别为5.42和7.24。pH≤3.4时微溶,pH>3.4时,吉非替尼溶解度迅速下降,pH>6时,几乎不溶。因而选择在介质pH 1.0盐酸溶液、pH 4.5醋酸缓冲液、2 %吐温80水溶液和含2 %吐温80的pH 6.8磷酸盐缓冲液中评价自制片和对照片的溶出相似性。
根据《普通口服固体制剂溶出度试验技术指导原则》[5]配制介质pH 1.0盐酸溶液、pH 4.5醋酸缓冲液、含2 %吐温80的pH 6.8磷酸盐缓冲液、2 %吐温80水溶液。
2.3.2 对照品溶液的配制 称取吉非替尼对照品约25 mg,置入50 ml量瓶,加甲醇10 ml溶解,再加溶出介质稀释并定容成每1 ml含10 µg左右的溶液,备用。
2.3.3 测定波长的选择,取2.3.2项下溶液,配制一定浓度的吉非替尼溶液,用紫外分光光度计在200~400 nm范围内扫描,吉非替尼的最大吸收波长分别为340 nm(pH 1.0盐酸溶液、pH 4.5醋酸缓冲液)、334 nm(含2 %吐温80的pH 6.8磷酸盐缓冲液)、335 nm(2 %吐温80水溶液)
2.3.4 标准曲线的绘制 精密称定吉非替尼对照品25 mg,置入100 ml量瓶,用甲醇10 ml溶解,再用溶出介质分别稀释到刻度,得到浓度为250µg/ml的贮备液。分别用介质pH 1.0盐酸溶液、pH 4.5醋酸缓冲液、含2 %吐温80的pH 6.8磷酸盐缓冲液、2 %吐温80水溶液稀释成浓度为3,5,10,15,20,25 µg/ml的溶液,分别在340,340,334,335 nm处测定吸光度,以浓度C对相应的吸光度A进行线性回归,得到回归方程,4种介质的回归方程分别为A=0.035C-0.0056(r=0.9998),A=0.045C-0.0056(r=0.9997),A=0.041C-0.0078(r=0.9998),A=0.044C-0.0081(r=0.9999),表明在浓度3~25 µg/ml之间线性关系良好。
2.3.5 精密度试验 取上述2.3.4项下浓度 10 µg/ml的对照品溶液,在相应波长下测定吸光度,连续测定6次,在4种介质中测定的RSD值分别为0.51 %,0.63 %,0.71 %,0.65 %,均≤2 %,说明该测定方法的精密度良好。
2.3.6 稳定性试验 取2.3.4项下浓度为 10 µg/ml的对照品溶液,分别于0,1,2,3,5,7,10 h在相应波长下测定吸光度,结果显示吉非替尼在4种介质中吸光度的RSD值分别为0.35 %,0.43 %,0.51 %,0.47 %,表明样品在10 h内稳定。
2.3.7 回收率试验 称取处方量的辅料,加入一定量的吉非替尼对照药,配置成含药量高、中、低3个浓度的处方。分别采用4种介质配制成相应浓度的样品溶液,在4种介质中的回收率分别为99.45 %(RSD=0.51 %),99.12 %(RSD=0.55 %),98.45 %(RSD=0.67 %),99.34 %(RSD=0.78 %),表明药物在4种介质中回收率良好。
2.4 关键工艺的研究
2.4.1 粉碎 吉非替尼原料药是BCS 2类,溶解度低,在水中几乎不溶,粒度大小会对溶出度有影响,根据Ostwald-Freundli-ch方程,减少粒径能增加溶解度,加快溶出速率。主要采用分样筛筛分、万能粉碎(100目筛)、气流粉碎3种方式来处理原料药,不同粉碎分式得到的粒度结果见表1。
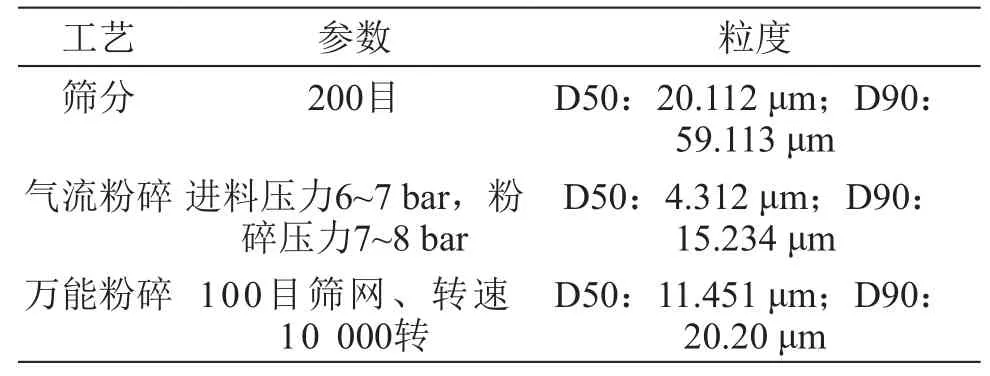
表1 原料药粒度分布
对表1数据进行分析,筛分得到的粒径明显太粗,不适合后续研究。粒径越小越能促进溶出,气流粉碎得到的粒径最小,但气流粉碎过程耗时,损失原料多。虽然万能粉碎得到的粒径稍大于气流粉碎,但考虑到万能粉碎工艺简单,损失少,综合考虑最终选择万能粉碎。
2.4.2 混合 原辅料的混合对于含量的均匀度非常重要,为了保证混合工序中原辅料混合均匀,固定设备搅拌速度,测定不同混合时间下的混合均匀度(RSD值)。称取处方量的吉非替尼、一水乳糖、微晶纤维素、交联羧甲基纤维素钠等加入高效湿法混合制粒机中,混合,在6个不同位置(上、中、下)取样,紫外分光光度法测原料药含量,计算RSD值,结果见表2。

表2 混合时间及均匀度
由表2可见,在设定的转速下,原、辅料在湿法混合制粒机中混合 5 min,即可实现混合均匀(RSD<2 %)。为了确保物料能充分混合均匀,将该混合工艺的混合时间定为10 min。
2.4.3 干燥时间 湿颗粒在60 ℃下鼓风干燥,每30 min翻盘一次,分别烘干30,60,90,120 min,采用水分测定仪,温度105 ℃,时间10 min,分别测定水分,结果见表3。

表3 小试3批颗粒水分测定结果
根据上述实验结果,干燥温度55~65 ℃,干燥90 min后颗粒水分可以满足压片的需要,干燥60 min水分偏大,干燥2 h水分太少,也不利于压片。所以最终选择的干燥时间为90 min。
2.5 溶出曲线评价[6]
参照中国药典2015版0931第二法制订了溶出度测定方法。分别采用介质pH 1.0盐酸溶液、pH 4.5醋酸缓冲液、含2 %吐温80的pH 6.8磷酸盐缓冲液、2 %吐温80水溶液各900 ml为溶出介质,溶出介质经脱气处理,转速为50 r/min,温度37 ℃,依法操作,分别于5,10,15,30,45,60 min取样10 ml,并及时补液,滤过后,取续滤液分别用4种介质稀释成每1 ml中含吉非替尼10 μg的溶液,摇匀,作为供试品溶液;另取吉非替尼对照品约25 mg,精密称定,置入50 ml量瓶,加甲醇10 ml溶解,加4种介质稀释至刻度,摇匀,精密量取适量,分别加4种介质并稀释成每1 ml中约含吉非替尼10 μg的溶液,作为对照品溶液。取供试品溶液和对照品溶液,照紫外-可见分光光度法[7],分别测定吸光度,计算每个点的溶出量。
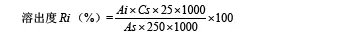

式中,As为对照品溶液的吸光度值,Ai为供试品溶液的吸光度值,Cs为对照品溶液浓度(μg/ml)。
按上述拟定的溶出度测定条件,分别测定自制制剂(批次为170201,170202,170203)与参比制剂在4种介质中的溶出度。见图1~4。

图1 吉非替尼片在pH 1.0盐酸溶液中的溶出曲线

图2 吉非替尼片在pH 4.5醋酸盐缓冲液中的溶出曲线

图3 吉非替尼片在2%吐温80水中的溶出曲线

图4 吉非替尼片在含2 %吐温80的pH 6.8磷酸盐缓冲液中的溶出曲线
从图1~4可见,自制制剂和参比制剂在pH 1.0和pH 4.5介质中15 min的溶出度达85 %以上,可认为两者在此溶出条件下溶出曲线相似。吉非替尼在水溶液中的溶解度具有pH依赖性,在pH 4~6之间,溶解度急剧下降,随pH值升高,溶解度降低,体外溶出减缓,参比片在水中和pH 6.8磷酸缓冲盐溶液中的溶出度很低,不足5 %,加入2 %吐温80后,溶出度明显升高。分别测定自制片和参比片在pH 6.8磷酸盐缓冲液(含2 %吐温80)和水溶液(含2 %吐温80)2种介质中的溶出曲线,按《仿制药生物等效性试验指导原则》[8],计算相似因子f2,其f2值分别为65.45,70.2,均大于50,可认为自制制剂和参比制剂在这2种介质中具有相似性。
2.6 样品加速考察后的溶出曲线研究
将制备的3批次样品(批号170201,170202,170203)和参比制剂置于40 ±2 ℃,相对湿度(RH)75%±5 %条件下加速考察6个月。分别在pH 1.0盐酸溶液、pH 4.5醋酸缓冲液、含2 %吐温80的pH 6.8磷酸盐缓冲液、2 %吐温80水溶液介质中测定溶出度,并计算f2值。结果见表4,表明制备的样品在加速考察后溶出曲线依然具有相似性。
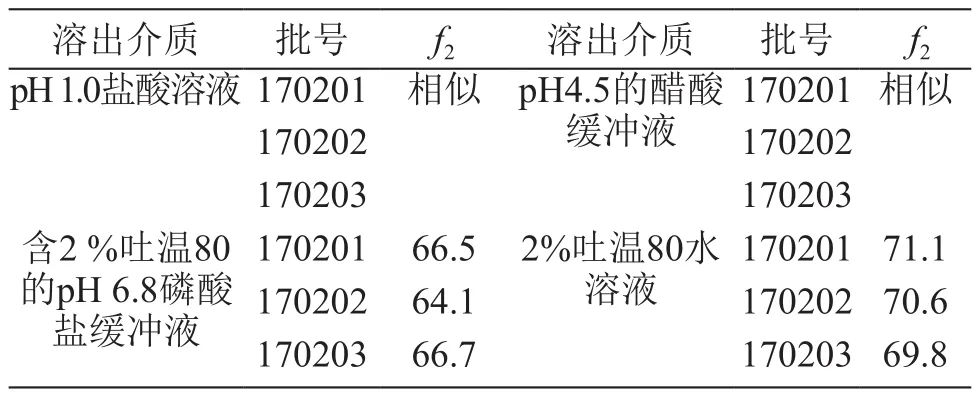
表4 吉非替尼片的溶出度结果
3 讨论
吉非替尼为难溶性药物,其粒度大小会对溶出度有影响,故对原料的粒度和粉碎方式进行了研究,最终确定采用万能粉碎机对原料药进行粉碎。吉非替尼原料含其无水物和三水合物两种晶型,制备过程中有可能转晶[9]。故制剂制备工艺应注意控制环境湿度和物料水分,所以对物料干燥时间进行了研究,最终选择的干燥时间为90 min。本研究采用常规湿法制粒压片方法制备吉非替尼片,通过对粒度、混合均匀度及干燥时间的研究,确定了关键工艺参数。由此工艺制备3个批次(每批次2000片),进行溶出曲线评价。
根据吉非替尼溶解度pH依赖性,且在pH 4~6之间溶解度急剧下降的特点,重点比较了pH 1.0盐酸溶液、pH 4.5醋酸缓冲液、含2 %吐温80的pH 6.8磷酸盐缓冲液、2 %吐温80水溶液4种介质中的溶出行为,4种介质中吉非替尼片溶出行为有明显差异,且具有一定代表性。数据显示,自制3批制剂与参比制剂在该4种介质中拟合度均高于50,说明二者溶出曲线具有相似性。其中吉非替尼在水中和磷酸6.8缓冲液中不溶,进口注册标准采用5 %吐温水溶液作为溶出介质,但45 min溶出即高于85 %,溶出略快。通过降低吐温用量至2 %,60~90 min间溶出达85 %以上,曲线平缓且溶出完全,具有一定区分度,所以采用了含2 %吐温80的pH 6.8磷酸盐缓冲液和2 %吐温80水溶液两种介质,结果表明自制3批制剂与参比制剂在该2种介质中溶出曲线具有相似性。
猜你喜欢
电子乐园·上旬刊(2022年5期)2022-04-09
快乐语文(2020年11期)2020-06-06
小学生学习指导(爆笑校园)(2020年3期)2020-06-05
天然产物研究与开发(2018年8期)2018-09-10
中成药(2018年1期)2018-02-02
中成药(2017年10期)2017-11-16
中成药(2017年10期)2017-11-16
阅读(中年级)(2016年9期)2016-05-14
特产研究(2016年3期)2016-04-12
中国药业(2014年17期)2014-05-26
